
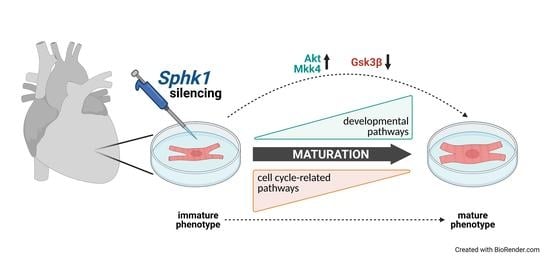
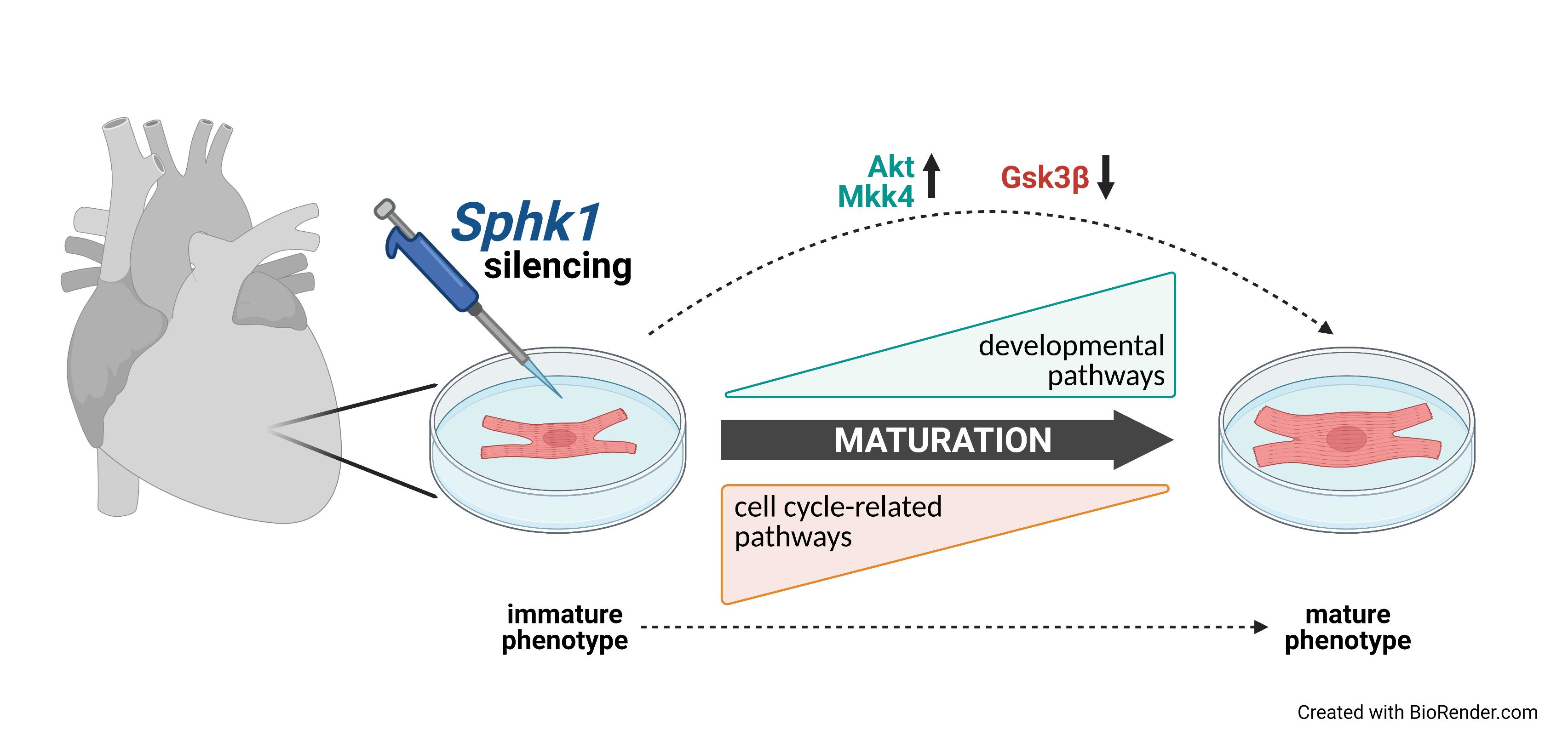
Silencing of Sphingosine kinase 1 Affects Maturation Pathways in Mouse Neonatal Cardiomyocytes

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
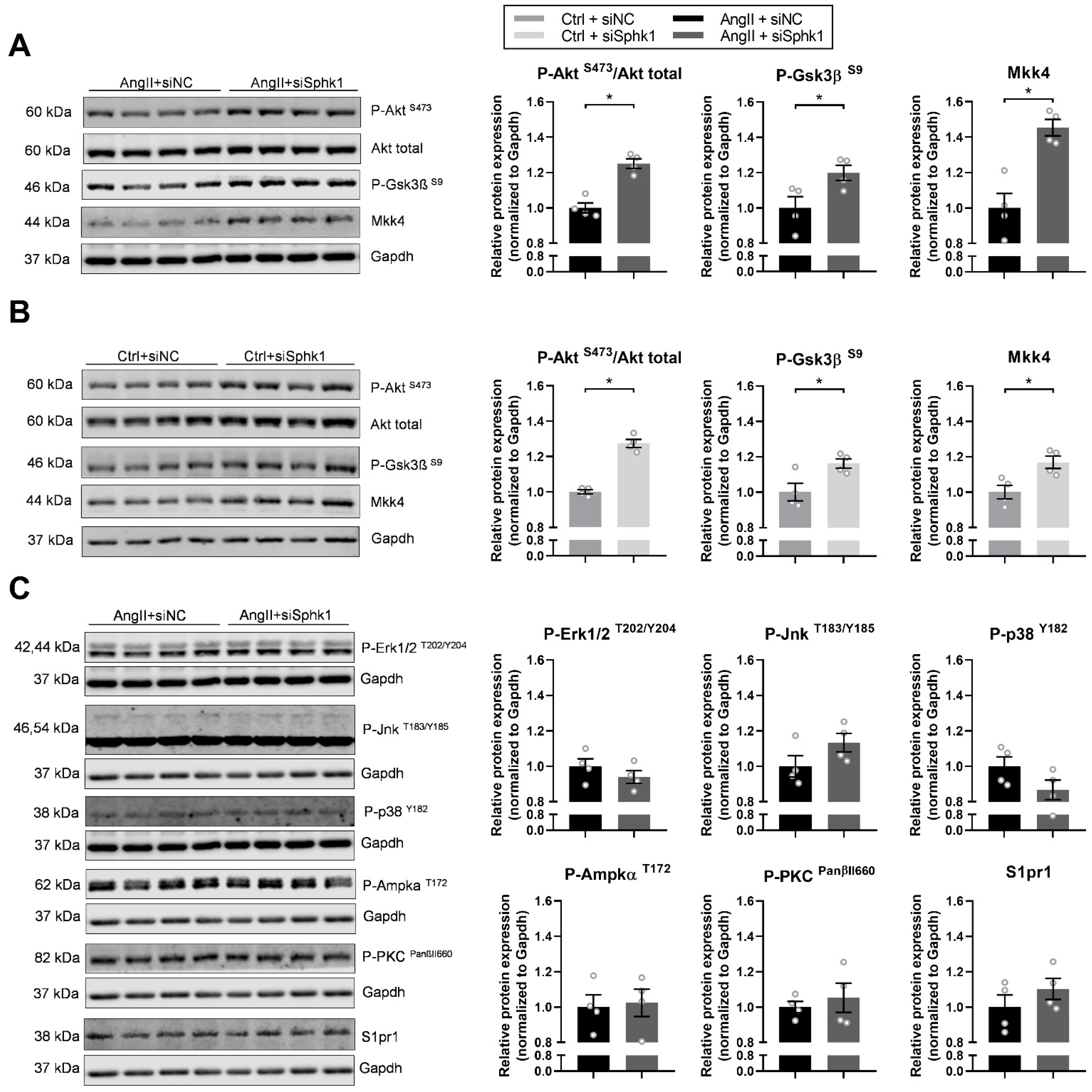
2. Results
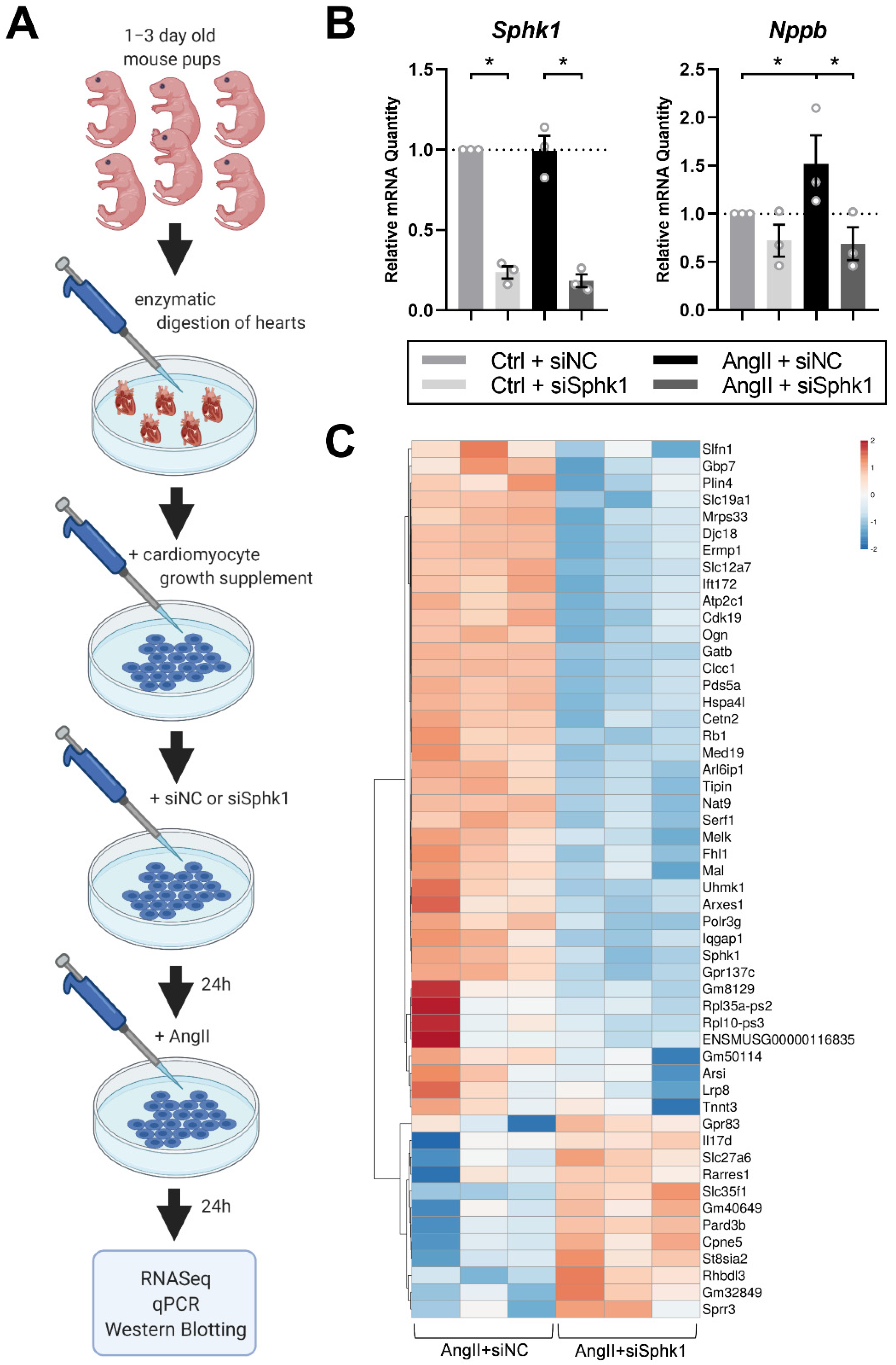
2.1. Sphk1 Silencing Affects the Neonatal CM Transcriptome in the Presence of AngII
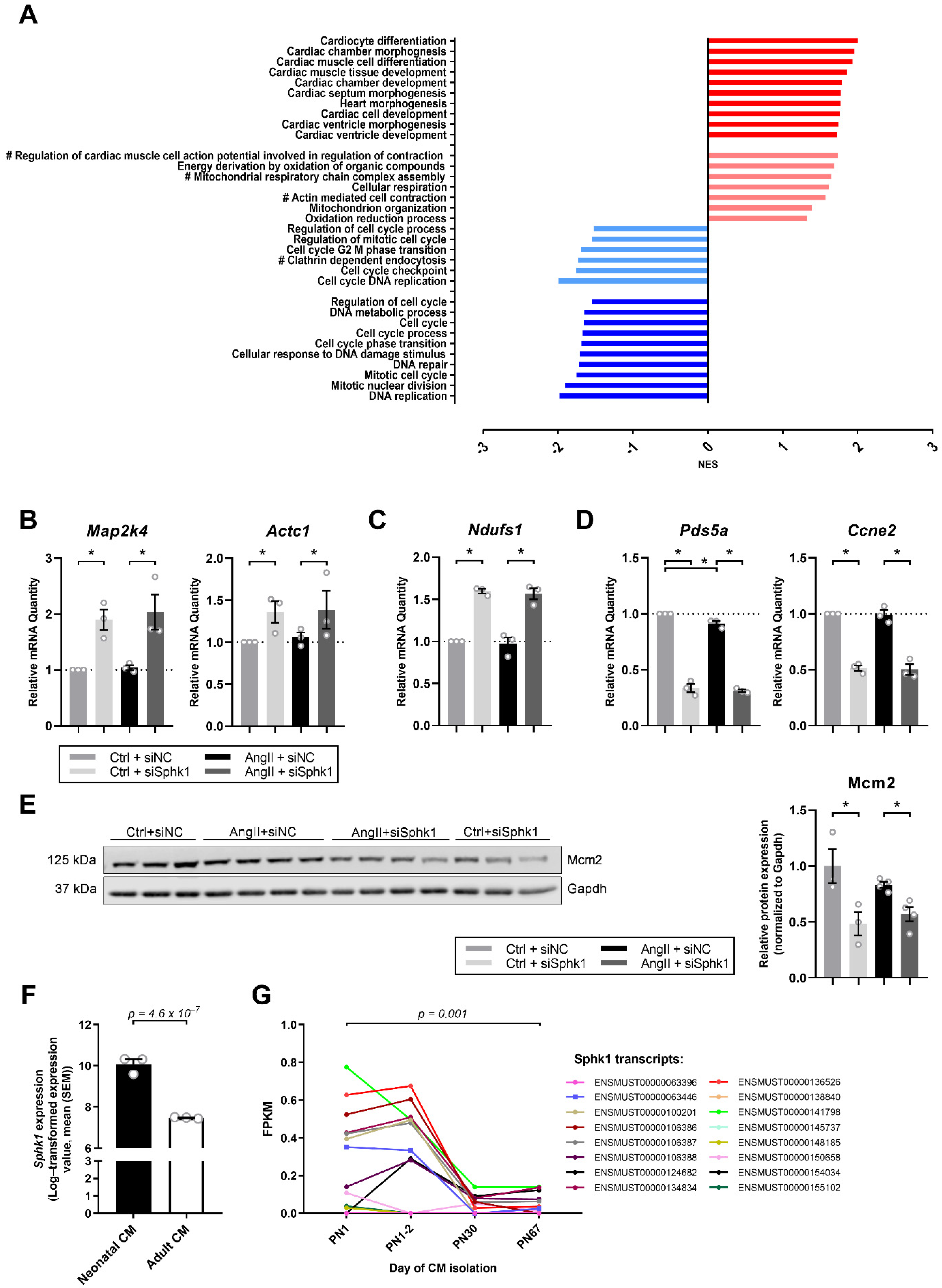
2.2. Sphk1 Silencing Influences Pathways Related to CM Development and Proliferation
2.3. Key Signaling Pathways Mediate the Effects of Sphk1 Silencing in Neonatal CMs
3. Discussion
4. Materials and Methods
4.1. Neonatal Mouse Cardiomyocytes
4.2. RNASeq Analysis
4.3. Real Time PCR
4.4. Analyses of Murine Sphk1 Expression in CMs over Time
4.5. Western Blotting
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sayers, J.R.; Riley, P.R. Heart regeneration: Beyond new muscle and vessels. Cardiovasc. Res. 2021, 117, 727–742. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Krysztofiak, H.; Petkow Dimitrow, P. Differentiating physiology from pathology in elite athletes. Left ventricular hypertrophy versus hypertrophic cardiomyopathy. Kardiol. Pol. 2016, 74, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Leinwand, L.A. Pregnancy as a cardiac stress model. Cardiovasc. Res. 2014, 101, 561–570. [Google Scholar] [CrossRef]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.P.; Yamashita, T.; Proia, R.L. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.; Wilsbacher, L.D.; Wilson, S.J.; Duong, D.N.; McDonald, M.; Lam, I.; Park, K.E.; Chun, J.; Coughlin, S.R. Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev. Biol. 2016, 418, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Belyea, B.C.; Li, M.; Gothert, J.R.; Gomez, R.A.; Sequeira-Lopez, M.L. Identification of cardiac hemo-vascular precursors and their requirement of sphingosine-1-phosphate receptor 1 for heart development. Sci. Rep. 2017, 7, 45205. [Google Scholar] [CrossRef]
- MacRitchie, N.; Volpert, G.; Al Washih, M.; Watson, D.G.; Futerman, A.H.; Kennedy, S.; Pyne, S.; Pyne, N.J. Effect of the sphingosine kinase 1 selective inhibitor, PF-543 on arterial and cardiac remodelling in a hypoxic model of pulmonary arterial hypertension. Cell Signal. 2016, 28, 946–955. [Google Scholar] [CrossRef][Green Version]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, S.-I.; Usui, S.; Takashima, S.-I.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Inagaki, Y.; Sugimoto, N.; Kitano, T.; Takamura, M.; et al. Augmented sphingosine 1 phosphate receptor-1 signaling in cardiac fibroblasts induces cardiac hypertrophy and fibrosis through angiotensin II and interleukin-6. PLoS ONE 2017, 12, e0182329. [Google Scholar] [CrossRef] [PubMed]
- Siedlinski, M.; Nosalski, R.; Szczepaniak, P.; Ludwig-Gałęzowska, A.H.; Mikołajczyk, T.; Filip, M.; Osmenda, G.; Wilk, G.; Nowak, M.; Wołkow, P.; et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci. Rep. 2017, 7, 44131. [Google Scholar] [CrossRef] [PubMed]
- Józefczuk, E.; Nosalski, R.; Saju, B.; Crespo, E.; Szczepaniak, P.; Guzik, T.J.; Siedlinski, M. Cardiovascular Effects of Pharmacological Targeting of Sphingosine Kinase 1. Hypertension 2020, 75, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Diniz, G.P.; Carneiro-Ramos, M.S.; Barreto-Chaves, M.L. Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3beta/mTOR signaling pathway. Basic Res. Cardiol. 2009, 104, 653–667. [Google Scholar] [CrossRef]
- Seo, K.; Parikh, V.N.; Ashley, E.A. Stretch-Induced Biased Signaling in Angiotensin II Type 1 and Apelin Receptors for the Mediation of Cardiac Contractility and Hypertrophy. Front. Physiol. 2020, 11, 181. [Google Scholar] [CrossRef]
- Gardner, D. Natriuretic peptides: Markers or modulators of cardiac hypertrophy? Trends Endocrinol. Metab. 2003, 14, 411–416. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Giudice, J.; Xia, Z.; Wang, E.T.; Scavuzzo, M.A.; Ward, A.J.; Kalsotra, A.; Wang, W.; Wehrens, X.H.; Burge, C.B.; Li, W.; et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 2014, 5, 3603. [Google Scholar] [CrossRef]
- Cattaneo, P.; Kunderfranco, P.; Greco, C.; Guffanti, A.; Stirparo, G.G.; Rusconi, F.; Rizzi, R.; Di Pasquale, E.; Locatelli, S.L.; Latronico, M.V.; et al. DOT1L-mediated H3K79me2 modification critically regulates gene expression during cardiomyocyte differentiation. Cell Death Differ. 2016, 23, 555–564. [Google Scholar] [CrossRef]
- Zhang, J.; Honbo, N.; Goetzl, E.J.; Chatterjee, K.; Karliner, J.S.; Gray, M.O. Signals from type 1 sphingosine 1-phosphate receptors enhance adult mouse cardiac myocyte survival during hypoxia. Am. J. Physiol. Circ. Physiol. 2007, 293, H3150–H3158. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.T.; Ye, S.; Su, J.; Garg, V. Cardiomyocyte Proliferation and Maturation: Two Sides of the Same Coin for Heart Regeneration. Front. Cell Dev. Biol 2020, 8, 594226. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Teppo, J.; Poho, P.; Movahedi, P.; Vaikkinen, A.; Karhu, S.T.; Trost, K.; Suvitaival, T.; Heikkonen, J.; Pahikkala, T.; et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Heart Assoc. 2018, 7, e010378. [Google Scholar] [CrossRef]
- Clement, S.; Stouffs, M.; Bettiol, E.; Kampf, S.; Krause, K.H.; Chaponnier, C.; Jaconi, M. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J. Cell Sci. 2007, 120 Pt 2, 229–238. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Ko, C.I.; Zhang, L.; Puga, A.; Xia, Y. Distinct signaling properties of mitogen-activated protein kinase kinases 4 (MKK4) and 7 (MKK7) in embryonic stem cell (ESC) differentiation. J. Biol. Chem. 2012, 287, 2787–2797. [Google Scholar] [CrossRef]
- Liu, W.; Zi, M.; Jin, J.; Prehar, S.; Oceandy, D.; Kimura, T.E.; Lei, M.; Neyses, L.; Weston, A.H.; Cartwright, E.J.; et al. Cardiac-specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ. Res. 2009, 104, 905–914. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Terret, M.E.; Sherwood, R.; Rahman, S.; Qin, J.; Jallepalli, P.V. Cohesin acetylation speeds the replication fork. Nature 2009, 462, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S.; Todorov, I.T.; Melendy, T.; Gilbert, D.M. Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J. Cell Biol. 1999, 146, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Kohama, T.; Edsall, L.; Nava, V.; Cuvillier, O.; Poulton, S.; Spiegel, S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 1999, 147, 545–558. [Google Scholar] [CrossRef]
- Shiojima, I.; Yefremashvili, M.; Luo, Z.; Kureishi, Y.; Takahashi, A.; Tao, J.; Rosenzweig, A.; Kahn, C.R.; Abel, E.D.; Walsh, K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 2002, 277, 37670–37677. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K. Akt signaling and growth of the heart. Circulation 2006, 113, 2032–2034. [Google Scholar] [CrossRef]
- Yan, Z.P.; Li, J.T.; Zeng, N.; Ni, G.X. Role of extracellular signal-regulated kinase 1/2 signaling underlying cardiac hypertrophy. Cardiol. J. 2020. [Google Scholar] [CrossRef]
- Mutlak, M.; Kehat, I. Extracellular signal-regulated kinases 1/2 as regulators of cardiac hypertrophy. Front. Pharmacol. 2015, 6, 149. [Google Scholar] [CrossRef]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. The complex network of mTOR signaling in the heart. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Michael, A.; Haq, S.; Chen, X.; Hsich, E.; Cui, L.; Walters, B.; Shao, Z.; Bhattacharya, K.; Kilter, H.; Huggins, G.; et al. Glycogen Synthase Kinase-3β Regulates Growth, Calcium Homeostasis, and Diastolic Function in the Heart. J. Biol. Chem. 2004, 279, 21383–21393. [Google Scholar] [CrossRef]
- Hirotani, S.; Zhai, P.; Tomita, H.; Galeotti, J.; Marquez, J.P.; Gao, S.; Hong, C.; Yatani, A.; Avila, J.; Sadoshima, J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ. Res. 2007, 101, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Bhattacharya, K.; Hsich, E.; Park, L.; Walters, B.; Germann, U.; Wang, Y.-M.; Kyriakis, J.; Mohanlal, R.; Kuida, K.; et al. c-Jun N-Terminal Kinases Mediate Reactivation of Akt and Cardiomyocyte Survival After Hypoxic Injury In Vitro and In Vivo. Circ. Res. 2006, 98, 111–118. [Google Scholar] [CrossRef]
- Dyck, J.R.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: Enemy or ally? J. Physiol. 2006, 574 Pt 1, 95–112. [Google Scholar] [CrossRef]
- Chan, A.Y.; Soltys, C.L.; Young, M.E.; Proud, C.G.; Dyck, J.R. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J. Biol. Chem. 2004, 279, 32771–32779. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.L.; Paquet, L.; Allen, B.G.; Rindt, H. Protein kinase C isoform expression and activity in the mouse heart. Am. J. Physiol. Circ. Physiol. 2001, 281, H2062–H2071. [Google Scholar] [CrossRef]
- Hamplova, B.; Novakova, O.; Tvrzicka, E.; Kolar, F.; Novak, F. Protein kinase C activity and isoform expression during early postnatal development of rat myocardium. Cell Biochem. Biophys. 2005, 43, 105–117. [Google Scholar] [CrossRef]
- Simon, J.N.; Chowdhury, S.A.; Warren, C.M.; Sadayappan, S.; Wieczorek, D.F.; Solaro, R.J.; Wolska, B.M. Ceramide-mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res. Cardiol. 2014, 109, 445. [Google Scholar] [CrossRef]
- Foldes, G.; Mioulane, M.; Wright, J.S.; Liu, A.Q.; Novak, P.; Merkely, B.; Gorelik, J.; Schneider, M.D.; Ali, N.N.; Harding, S.E. Modulation of human embryonic stem cell-derived cardiomyocyte growth: A testbed for studying human cardiac hypertrophy? J. Mol. Cell. Cardiol. 2011, 50, 367–376. [Google Scholar] [CrossRef]
- Wu, L.; Jia, Z.; Yan, L.; Wang, W.; Wang, J.; Zhang, Y.; Zhou, C. Angiotensin II promotes cardiac differentiation of embryonic stem cells via angiotensin type 1 receptor. Differentiation 2013, 86, 23–29. [Google Scholar] [CrossRef]
- Kemi, O.J.; Ceci, M.; Wisloff, U.; Grimaldi, S.; Gallo, P.; Smith, G.L.; Condorelli, G.; Ellingsen, O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J. Cell. Physiol. 2008, 214, 316–321. [Google Scholar] [CrossRef]
- Drenckhahn, J.D.; Strasen, J.; Heinecke, K.; Langner, P.; Yin, K.V.; Skole, F.; Hennig, M.; Spallek, B.; Fischer, R.; Blaschke, F.; et al. Impaired myocardial development resulting in neonatal cardiac hypoplasia alters postnatal growth and stress response in the heart. Cardiovasc. Res. 2015, 106, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ovics, P.; Regev, D.; Baskin, P.; Davidor, M.; Shemer, Y.; Neeman, S.; Ben-Haim, Y.; Binah, O. Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening. Int. J. Mol. Sci. 2020, 21, 7320. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, J.-D.; Fang, X.-H.; Zhu, J.-N.; Yang, J.; Pan, R.; Yuan, S.-J.; Zeng, N.; Yang, Z.-Z.; Yang, H.; et al. Circular RNA circRNA_000203 aggravates cardiac hypertrophy via suppressing miR-26b-5p and miR-140-3p binding to Gata4. Cardiovasc. Res. 2020, 116, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-M.; Liu, F.-Z.; Zhu, J.-N.; Fu, Y.-H.; Lin, Q.-X.; Deng, C.-Y.; Hu, Z.-Q.; Yang, H.; Zheng, X.-L.; Cheng, J.-D.; et al. Myocyte-specific enhancer factor 2C: A novel target gene of miR-214-3p in suppressing angiotensin II-induced cardiomyocyte hypertrophy. Sci. Rep. 2016, 6, 36146. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.M.; Rivera, P.D.; Lacagnina, M.J.; Hanamsagar, R.; Bilbo, S.D. Optimized solubilization of TRIzol-precipitated protein permits Western blotting analysis to maximize data available from brain tissue. J. Neurosci. Methods 2017, 280, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021, 060012. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jozefczuk, E.; Szczepaniak, P.; Guzik, T.J.; Siedlinski, M. Silencing of Sphingosine kinase 1 Affects Maturation Pathways in Mouse Neonatal Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 3616. https://doi.org/10.3390/ijms22073616
Jozefczuk E, Szczepaniak P, Guzik TJ, Siedlinski M. Silencing of Sphingosine kinase 1 Affects Maturation Pathways in Mouse Neonatal Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(7):3616. https://doi.org/10.3390/ijms22073616
Chicago/Turabian StyleJozefczuk, Ewelina, Piotr Szczepaniak, Tomasz Jan Guzik, and Mateusz Siedlinski. 2021. "Silencing of Sphingosine kinase 1 Affects Maturation Pathways in Mouse Neonatal Cardiomyocytes" International Journal of Molecular Sciences 22, no. 7: 3616. https://doi.org/10.3390/ijms22073616
APA StyleJozefczuk, E., Szczepaniak, P., Guzik, T. J., & Siedlinski, M. (2021). Silencing of Sphingosine kinase 1 Affects Maturation Pathways in Mouse Neonatal Cardiomyocytes. International Journal of Molecular Sciences, 22(7), 3616. https://doi.org/10.3390/ijms22073616