
Functional and Molecular Properties of DYT-SGCE Myoclonus-Dystonia Patient-Derived Striatal Medium Spiny Neurons

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Differentiation of iPSCs into Medium Spiny Neurons (MSNs)
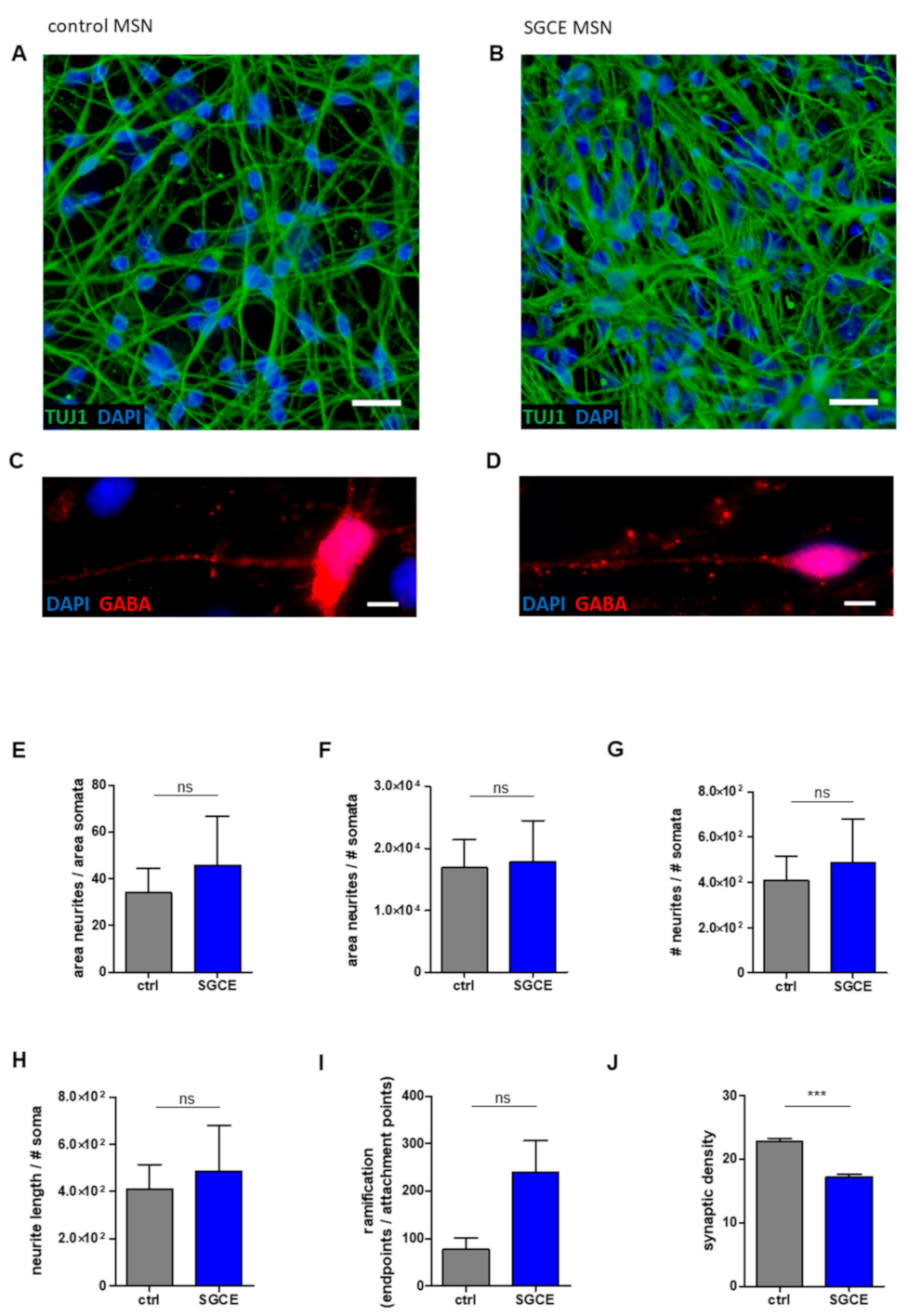
2.2. Reduced Synaptic Density in SGCE MSNs
2.3. Ca2+ Signaling in MSNs
2.3.1. Elevated Basal Intracellular Ca2+ Levels and Lower Frequency of Spontaneous Ca2+ Signals in SGCE MSNs
2.3.2. Elevated Ca2+ Amplitudes upon Glycine and Acetylcholine Application in SGCE MSNs
2.3.3. Blocking of Ca2+ Amplitudes after Application of Antagonists for Voltage-Gated Ca2+ Channels and Acetylcholine Receptors in MSNs
2.4. Expression of Voltage-Gated Ca2+ Channels and Ionotropic Receptor Subunits in MSNs
2.5. Properties of Voltage-Gated Ion Channels, Synaptic Activity and Action Potentials in MSNs
2.5.1. Larger mPSC Amplitudes in SGCE MSNs
2.5.2. Elevated Amplitudes of Evoked Action Potentials in SGCE MSNs
3. Discussion
4. Materials and Methods
4.1. Cultivation of Human iPSC Lines
4.2. Differentiation of iPSC Lines into Striatal Medium Spiny Neurons (MSNs)
4.3. Immunocytochemistry
4.4. Analysis of Neuronal Morphology and Synaptic Density
4.5. Calcium Imaging
4.6. Quantitative Real-Time PCR
4.7. Electrophysiology
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinugawa, K.; Vidailhet, M.; Clot, F.; Apartis, E.; Grabli, D.; Roze, E. Myoclonus-dystonia: An update. Mov. Disord. 2009, 24, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 2013, 28, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Peall, K.J.; Kurian, M.A.; Wardle, M.; Waite, A.J.; Hedderly, T.; Lin, J.-P.; Smith, M.; Whone, A.L.; Pall, H.; White, C.; et al. SGCE and myoclonus dystonia: Motor characteristics, diagnostic criteria and clinical predictors of genotype. J. Neurol. 2014, 261, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lang, A.; Van De Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef]
- Roze, E.; Lang, A.E.; Vidailhet, M. Myoclonus-dystonia: Classification, phenomenology, pathogenesis, and treatment. Curr. Opin. Neurol. 2018, 31, 484–490. [Google Scholar] [CrossRef]
- Asmus, F.; Zimprich, A.; Du Montcel, S.T.; Kabus, C.; Deuschl, G.; Kupsch, A.; Ziemann, U.; Castro, M.; Kühn, A.A.; Strom, T.M.; et al. Myoclonus-dystonia syndrome: ε-sarcoglycan mutations and phenotype. Ann. Neurol. 2002, 52, 489–492. [Google Scholar] [CrossRef]
- Saunders–Pullman, R.; Shriberg, J.; Heiman, G.; Raymond, D.; Wendt, K.; Kramer, P.; Schilling, K.; Kurlan, R.; Klein, C.; Ozelius, L.J.; et al. Myoclonus dystonia: Possible association with obsessive-compulsive disorder and alcohol dependence. Neurology 2002, 58, 242–245. [Google Scholar] [CrossRef]
- Hess, C.W.; Raymond, D.; Aguiar, P.D.C.; Frucht, S.; Shriberg, J.; Heiman, G.A.; Kurlan, R.; Klein, C.; Bressman, S.B.; Ozelius, L.J.; et al. Myoclonus-dystonia, obsessive-compulsive disorder, and alcohol dependence in SGCE mutation carriers. Neurology 2007, 68, 522–524. [Google Scholar] [CrossRef]
- Weissbach, A.; Kasten, M.; Grünewald, A.; Brüggemann, N.; Trillenberg, P.; Klein, C.; Hagenah, J. Prominent psychiatric comorbidity in the dominantly inherited movement disorder myoclonus-dystonia. Park. Relat. Disord. 2013, 19, 422–425. [Google Scholar] [CrossRef]
- Peall, K.J.; Dijk, J.M.; Saunders-Pullman, R.; Dreissen, Y.E.M.; Van Loon, I.; Cath, D.C.; Kurian, M.A.; Owen, M.J.; Foncke, E.M.J.; Morris, H.R.; et al. Psychiatric disorders, myoclonus dystonia and SGCE: An international study. Ann. Clin. Transl. Neurol. 2016, 3, 4–11. [Google Scholar] [CrossRef]
- Cif, L.; Valente, E.M.; Hemm, S.; Coubes, C.; Vayssiere, N.; Serrat, S.; Di Giorgio, A.; Coubes, P. Deep brain stimulation in myoclonus-dystonia syndrome. Mov. Disord. 2004, 19, 724–727. [Google Scholar] [CrossRef]
- Kurtis, M.M.; Luciano, M.S.; Yu, Q.; Goodman, R.R.; Ford, B.; Raymond, D.; Pullman, S.L.; Saunders-Pullman, R. Clinical and neurophysiological improvement of SGCE myoclonus–dystonia with GPi deep brain stimulation. Clin. Neurol. Neurosurg. 2010, 112, 149–152. [Google Scholar] [CrossRef]
- Thenganatt, M.A.; Jankovic, J. Treatment of Dystonia. Neurotherapeutics 2013, 11, 139–152. [Google Scholar] [CrossRef]
- Zimprich, A.; Grabowski, M.; Asmus, F.; Naumann, M.; Berg, D.; Bertram, M.; Scheidtmann, K.H.; Kern, P.; Winkelmann, J.; Müller-Myhsok, B.; et al. Mutations in the gene encoding ɛ-sarcoglycan cause myoclonus–dystonia syndrome. Nat. Genet. 2001, 29, 66–69. [Google Scholar] [CrossRef]
- Asmus, F.; Salih, F.; Hjermind, L.E.; Ostergaard, K.; Munz, M.; Kühn, A.A.; Dupont, E.; Kupsch, A.; Gasser, T. Myoclonus-dystonia due to genomic deletions in the epsilon-sarcoglycan gene. Ann. Neurol. 2005, 58, 792–797. [Google Scholar] [CrossRef]
- Grünewald, A.; Djarmati, A.; Lohmann-Hedrich, K.; Farrell, K.; Zeller, J.A.; Allert, N.; Papengut, F.; Petersen, B.; Fung, V.; Sue, C.M.; et al. Myoclonus-dystonia: Significance of large SGCE deletions. Hum. Mutat. 2008, 29, 331–332. [Google Scholar] [CrossRef]
- Grabowski, M.; Zimprich, A.; Lorenz-Depiereux, B.; Kalscheuer, V.; Asmus, F.; Gasser, T.; Meitinger, T.; Strom, T.M. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur. J. Hum. Genet. 2003, 11, 138–144. [Google Scholar] [CrossRef]
- Müller, B.; Hedrich, K.; Kock, N.; Dragasevic, N.; Svetel, M.; Garrels, J.; Landt, O.; Nitschke, M.; Pramstaller, P.P.; Reik, W.; et al. Evidence That Paternal Expression of the ε-Sarcoglycan Gene Accounts for Reduced Penetrance in Myoclonus-Dystonia. Am. J. Hum. Genet. 2002, 71, 1303–1311. [Google Scholar] [CrossRef]
- Grütz, K.; Seibler, P.; Weissbach, A.; Lohmann, K.; Carlisle, F.A.; Blake, D.J.; Westenberger, A.; Klein, C.; Grünewald, A. Faithful SGCE imprinting in iPSC-derived cortical neurons: An endogenous cellular model of myoclonus-dystonia. Sci. Rep. 2017, 7, srep41156. [Google Scholar] [CrossRef]
- Ozawa, E.; Mizuno, Y.; Hagiwara, Y.; Sasaoka, T.; Yoshida, M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005, 32, 563–576. [Google Scholar] [CrossRef]
- Ritz, K.; van Schaik, B.D.; Jakobs, M.E.; van Kampen, A.H.; Aronica, E.; Tijssen, M.A.; Baas, F. SGCE isoform characterization and expression in human brain: Implications for myoclonus-dystonia pathogenesis? Eur. J. Hum. Genet. 2011, 19, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.J.; Carlisle, F.A.; Chan, Y.M.; Blake, D.J. Myoclonus dystonia and muscular dystrophy: ɛ-sarcoglycan is part of the dystrophin-associated protein complex in brain. Mov. Disord. 2016, 31, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Menozzi, E.; Balint, B.; Latorre, A.; Valente, E.M.; Rothwell, J.C.; Bhatia, K.P. Twenty years on: Myoclonus-dystonia and ε-sarcoglycan—Neurodevelopment, channel, and signaling dysfunction. Mov. Disord. 2019, 34, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, E.; Poetschke, C.; Duda, J.; Schlaudraff, F.; Lammel, S.; Schiemann, J.; Fauler, M.; Hetzel, A.; Watanabe, M.; Lujan, R.; et al. Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 2014, 137, 2287–2302. [Google Scholar] [CrossRef]
- Beukers, R.J.; Booij, J.; Weisscher, N.; Zijlstra, F.S.M.; Van Amelsvoort, T.A.M.J.; Tijssen, M.A.J. Reduced striatal D2 receptor binding in myoclonus–dystonia. Eur. J. Nucl. Med. Mol. Imaging 2008, 36, 269–274. [Google Scholar] [CrossRef][Green Version]
- Zhang, L.; Yokoi, F.; Parsons, D.S.; Standaert, D.G.; Li, Y. Alteration of Striatal Dopaminergic Neurotransmission in a Mouse Model of DYT11 Myoclonus-Dystonia. PLoS ONE 2012, 7, e33669. [Google Scholar] [CrossRef]
- Yokoi, F.; Dang, M.T.; Li, J.; Li, Y. Myoclonus, Motor Deficits, Alterations in Emotional Responses and Monoamine Metabolism in ε-Sarcoglycan Deficient Mice. J. Biochem. 2006, 140, 141–146. [Google Scholar] [CrossRef]
- Gasser, T. Inherited myoclonus-dystonia syndrome. Adv. Neurol. 1998, 78, 325–334. [Google Scholar]
- Weissbach, A.; Werner, E.; Bally, J.F.; Tunc, S.; Löns, S.; Timmann, D.; Zeuner, K.E.; Tadic, V.; Brüggemann, N.; Lang, A.; et al. Alcohol improves cerebellar learning deficit in myoclonus-dystonia: A clinical and electrophysiological investigation. Ann. Neurol. 2017, 82, 543–553. [Google Scholar] [CrossRef]
- Knuesel, I.; Mastrocola, M.; Zuellig, R.A.; Bornhauser, B.; Schaub, M.C.; Fritschy, J.-M. Altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur. J. Neurosci. 1999, 11, 4457–4462. [Google Scholar] [CrossRef]
- Kueh, S.L.L.; Head, S.I.; Morley, J.W. GABAAReceptor Expression and Inhibitory Post-Synaptic Currents in Cerebellar Purkinje Cells in Dystrophin-Deficient mdx Mice. Clin. Exp. Pharmacol. Physiol. 2007, 35, 207–210. [Google Scholar] [CrossRef]
- Waite, A.; Brown, S.C.; Blake, D.J. The dystrophin–glycoprotein complex in brain development and disease. Trends Neurosci. 2012, 35, 487–496. [Google Scholar] [CrossRef]
- Maltese, M.; Martella, G.; Imbriani, P.; Schuermans, J.; Billion, K.; Sciamanna, G.; Farook, F.; Ponterio, G.; Tassone, A.; Santoro, M.; et al. Abnormal striatal plasticity in a DYT11/SGCE myoclonus dystonia mouse model is reversed by adenosine A2A receptor inhibition. Neurobiol. Dis. 2017, 108, 128–139. [Google Scholar] [CrossRef]
- Yokoi, F.; Dang, M.T.; Zhou, T.; Li, Y. Abnormal nuclear envelopes in the striatum and motor deficits in DYT11 myoclonus-dystonia mouse models. Hum. Mol. Genet. 2011, 21, 916–925. [Google Scholar] [CrossRef]
- Washburn, S.; Fremont, R.; Moreno-Escobar, M.C.; Angueyra, C.; Khodakhah, K. Acute cerebellar knockdown of Sgce reproduces salient features of myoclonus-dystonia (DYT11) in mice. eLife 2019, 8. [Google Scholar] [CrossRef]
- Van Der Meer, J.N.; Beukers, R.J.; Van Der Salm, S.M.A.; Caan, M.W.A.; Tijssen, M.A.J.; Nederveen, A.J. White matter abnormalities in gene-positive myoclonus-dystonia. Mov. Disord. 2012, 27, 1666–1672. [Google Scholar] [CrossRef]
- Stanslowsky, N.; Reinhardt, P.; Glass, H.; Kalmbach, N.; Naujock, M.; Hensel, N.; Lübben, V.; Pal, A.; Venneri, A.; Lupo, F.; et al. Neuronal Dysfunction in iPSC-Derived Medium Spiny Neurons from Chorea-Acanthocytosis Patients Is Reversed by Src Kinase Inhibition and F-Actin Stabilization. J. Neurosci. 2016, 36, 12027–12043. [Google Scholar] [CrossRef]
- Capetian, P.; Stanslowsky, N.; Bernhardi, E.; Grütz, K.; Domingo, A.; Brüggemann, N.; Naujock, M.; Seibler, P.; Klein, C.; Wegner, F. Altered glutamate response and calcium dynamics in iPSC-derived striatal neurons from XDP patients. Exp. Neurol. 2018, 308, 47–58. [Google Scholar] [CrossRef]
- Haws, C.M.; Lansman, J.B. Calcium-permeable ion channels in cerebellar neurons from mdx mice. Proc. R. Soc. B Boil. Sci. 1991, 244, 185–189. [Google Scholar] [CrossRef]
- Hicks, C.B.; Abraham, K. Verapamil and Myoclonic Dystonia. Ann. Intern. Med. 1985, 103, 154. [Google Scholar] [CrossRef]
- Vadlamudi, L.; Wijdicks, E.F. Multifocal myoclonus due to verapamil overdose. Neurology 2002, 58, 984. [Google Scholar] [CrossRef]
- Goetz, C.G.; Horn, S.S. Treatment of tremor and dystonia. Neurol. Clin. 2001, 19, 129–144. [Google Scholar] [CrossRef]
- Raymond, D.; Saunders-Pullman, R.; Ozelius, L.A. SGCE Myoclonus-Dystonia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Jaunarajs, K.E.; Bonsi, P.; Chesselet, M.; Standaert, D.; Pisani, A. Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog. Neurobiol. 2015, 127-128, 91–107. [Google Scholar] [CrossRef]
- Fearon, C.; Peall, K.J.; Vidailhet, M.; Fasano, A. Medical management of myoclonus-dystonia and implications for underlying pathophysiology. Park. Relat. Disord. 2020, 77, 48–56. [Google Scholar] [CrossRef]
- Maltese, M.; Martella, G.; Madeo, G.; Fagiolo, I.; Tassone, A.; Ponterio, G.; Sciamanna, G.; Burbaud, P.; Conn, P.J.; Bonsi, P.; et al. Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: Role of M1muscarinic receptors. Mov. Disord. 2014, 29, 1655–1665. [Google Scholar] [CrossRef]
- Erosa-Rivero, H.B.; Bata-García, J.L.; Alvarez-Cervera, F.J.; Heredia-López, F.J.; Góngora-Alfaro, J.L. The potency and efficacy of anticholinergics to inhibit haloperidol-induced catalepsy in rats correlates with their rank order of affinities for the muscarinic receptor subtypes. Neuropharmacology 2014, 81, 176–187. [Google Scholar] [CrossRef]
- Hamann, M.; Plank, J.; Richter, F.; Bode, C.; Smiljanic, S.; Creed, M.; Nobrega, J.N.; Richter, A. Alterations of M1 and M4 acetylcholine receptors in the genetically dystonic (dtsz) hamster and moderate antidystonic efficacy of M1 and M4 anticholinergics. Neuroscience 2017, 357, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gutekunst, C.A.; Rees, H.D.; Heilman, C.J.; Levey, A.I. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: Light and electron microscopic immunocytochemistry using subtype- specific antibodies. J. Neurosci. 1994, 14, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Chatzidaki, A.; Fouillet, A.; Li, J.; Dage, J.; Millar, N.S.; Sher, E.; Ursu, D. Pharmacological Characterisation of Nicotinic Acetylcholine Receptors Expressed in Human iPSC-Derived Neurons. PLoS ONE 2015, 10, e0125116. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Moretti, M.; Zanardi, A.; McIntosh, J.M.; Clementi, F.; Gotti, C. Identification of the Nicotinic Receptor Subtypes Expressed on Dopaminergic Terminals in the Rat Striatum. J. Neurosci. 2002, 22, 8785–8789. [Google Scholar] [CrossRef] [PubMed]
- Ghedini, P.C.; Avellar, M.C.W.; De Lima, T.C.M.; Lima-Landman, M.T.R.; Lapa, A.J.; Souccar, C. Quantitative changes of nicotinic receptors in the hippocampus of dystrophin-deficient mice. Brain Res. 2012, 1483, 96–104. [Google Scholar] [CrossRef]
- Parames, S.; Coletta-Yudice, E.; Nogueira, F.; De Sousa, M.N.; Hayashi, M.; Lima-Landman, M.; Lapa, A.; Souccar, C. Altered acetylcholine release in the hippocampus of dystrophin-deficient mice. Neuroscience 2014, 269, 173–183. [Google Scholar] [CrossRef]
- Gruber, D.; Kühn, A.A.; Schoenecker, T.; Kivi, A.; Trottenberg, T.; Hoffmann, K.-T.; Gharabaghi, A.; Kopp, U.A.; Schneider, G.-H.; Klein, C.; et al. Pallidal and thalamic deep brain stimulation in myoclonus-dystonia. Mov. Disord. 2010, 25, 1733–1743. [Google Scholar] [CrossRef]
- Wang, J.-W.; Li, J.-P.; Wang, Y.-P.; Zhang, X.-H.; Zhang, Y.-Q. Deep brain stimulation for myoclonus-dystonia syndrome with double mutations in DYT1 and DYT11. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Krause, P.; Koch, K.; Gruber, D.; Kupsch, A.; Gharabaghi, A.; Schneider, G.; Kühn, A.A. Long-term effects of pallidal and thalamic deep brain stimulation in myoclonus dystonia. Eur. J. Neurol. 2021. [Google Scholar] [CrossRef]
- Japtok, J.; Lojewski, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef]
- Glaß, H.; Pal, A.; Reinhardt, P.; Sterneckert, J.; Wegner, F.; Storch, A.; Hermann, A. Defective mitochondrial and lysosomal trafficking in chorea-acanthocytosis is independent of Src-kinase signaling. Mol. Cell. Neurosci. 2018, 92, 137–148. [Google Scholar] [CrossRef]
- Staege, S.; Kutschenko, A.; Baumann, H.; Glaß, H.; Henkel, L.; Gschwendtberger, T.; Kalmbach, N.; Klietz, M.; Hermann, A.; Lohmann, K.; et al. Reduced expression of GABA(A) receptor alpha2 subunit is associated with disinhibition of DYT-THAP1 dystonia patient-derived striatal medium spiny neurons. Front. Cell. Dev. Biol. 2021. under review. [Google Scholar]
- WiCell. Feeder-independent culture protocol—EDTA splitting. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2014. [Google Scholar] [CrossRef]
- Ho, S.-Y.; Chao, C.-Y.; Huang, H.-L.; Chiu, T.-W.; Charoenkwan, P.; Hwang, E. NeurphologyJ: An automatic neuronal morphology quantification method and its application in pharmacological discovery. BMC Bioinform. 2011, 12, 1–18. [Google Scholar] [CrossRef]
- Meijering, E.; Jacob, M.; Sarria, J.-C.; Steiner, P.; Hirling, H.; Unser, M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytomatics 2004, 58, 167–176. [Google Scholar] [CrossRef]
- Hensel, N.; Schön, A.; Konen, T.; Lübben, V.; Förthmann, B.; Baron, O.; Grothe, C.; Leifheit-Nestler, M.; Claus, P.; Haffner, D. Fibroblast growth factor 23 signaling in hippocampal cells: Impact on neuronal morphology and synaptic density. J. Neurochem. 2016, 137, 756–769. [Google Scholar] [CrossRef]
- Stanslowsky, N.; Haase, A.; Martin, U.; Naujock, M.; Leffler, A.; Dengler, R.; Wegner, F. Functional differentiation of midbrain neurons from human cord blood-derived induced pluripotent stem cells. Stem Cell Res. Ther. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Wegner, F.; Kraft, R.; Busse, K.; Härtig, W.; Ahrens, J.; Leffler, A.; Dengler, R.; Schwarz, J. Differentiated Human Midbrain-Derived Neural Progenitor Cells Express Excitatory Strychnine-Sensitive Glycine Receptors Containing α2β Subunits. PLoS ONE 2012, 7, e36946. [Google Scholar] [CrossRef]
- Wegner, F.; Kraft, R.; Busse, K.; Härtig, W.; Schaarschmidt, G.; Schwarz, S.C.; Schwarz, J.; Hevers, W. Functional and molecular analysis of GABAA receptors in human midbrain-derived neural progenitor cells. J. Neurochem. 2008, 107, 1056–1069. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | DYT-SGCE | |||||
|---|---|---|---|---|---|---|
| Functional Properties | Control_1 (n = 21) | Control_2 (n = 14) | Control (n = 35) | SGCE_1 (n = 24) | SGCE_2 (n = 19) | SGCE (n = 43) |
| INa max. amplitudes (pA/pF) | −106.2 ± 18.8 | −126.6 ± 19.5 | −116.4 ± 10.2 | −85.75 ± 12.3 | −92.78 ± 14.72 | −89.27 ± 3.52 |
| IK max. amplitudes (pA/pF) | 148.4 ± 30.2 | 125.3 ± 26.3 | 136.9 ± 11.5 | 103.8 ± 13.1 | 128.4 ± 19.3 | 116.1 ± 12.3 |
| Resting membrane potential (mV) | −39.1 ± 2.2 | −23.3 ± 3.1 | −32.9 ± 2.2 | −33.4 ± 1.8 | −38.33 ± 3.0 | −35.7 ± 1.7 |
| Membrane capacitance (pF) | 30.5 ± 3.9 | 17.8 ± 3.1 | 25.4 ± 2.8 | 27.8 ± 4.6 | 28.2 ± 5.1 | 28.0 ± 3.4 |
| Input resistance (MOhm) | 996.3 ± 275.2 | 553.9 ± 195.2 | 819.4 ± 184.2 | 415.1 ± 74.6 | 566.1 ± 82.1 | 481.8 ± 55.8 |
| Cells with single evoked APs (%) | 48.9 ± 24.8 | 87.5 ± 12.5 | 68.2 ± 15.1 | 68.6 ± 2.0 | 72.2 ± 14.7 | 70.8 ± 8.1 |
| Cells with repetitive evoked APs (%) | 51.1 ± 24.8 | 12.5 ± 12.5 | 31.8 ± 15.1 | 31.4 ± 2.0 | 22.2 ± 14.7 | 25.9 ± 8.4 |
| Amplitude (mV) of evoked APs | 56.9 ± 5.1 | 57.8 ± 5.0 | 57.2 ± 3.7 | 75.8 ± 3.4 | 72.5 ± 4.1 | 74.3 ± 2.6 *** |
| Duration (ms) of evoked APs | 3.0 ± 0.4 | 3.5 ± 0.7 | 3.2 ± 0.4 | 2.4 ± 0.2 | 2.3 ± 0.1 | 2.3 ± 0.1 * |
| AHP amplitude (mV) | 7.6± 1.9 | 3.2 ± 0.7 | 6.0 ± 1.3 | 8.3 ± 1.1 | 8.1 ± 1.4 | 8.2 ± 0.9 * |
| Time to peak AHP (ms) | 21.6 ± 4.0 | 19.5 ± 3.0 | 20.9 ± 2.7 | 21.3 ± 2.0 | 31.9 ± 6.4 | 26.0 ± 3.1 |
| Cells with spontaneous APs (%) | 58.5 ± 20.3 | 37.5 ± 19.1 | 48.0 ± 13.3 | 74.5 ± 7.8 | 54.2 ± 23.2 | 62.3 ± 13.9 |
| Frequency of spontaneous APs (Hz) | 0.50 ± 0.10 | 0.47 ± 0.10 | 0.49 ± 0.07 | 0.52 ± 0.12 | 0.72 ± 0.16 | 0.60 ± 0.10 |
| Amplitude of spontaneous APs (mV) | 49.0 ± 3.1 | 42.7 ± 6.7 | 46.5 ± 3.2 | 46.7 ± 2.9 | 38.9 ± 3.4 | 43.3 ± 2.3 |
| Cells with miniature PSCs (%) | 96.3 ± 3.7 | 100.0 ± 0.0 | 98.2 ± 1.9 | 75.0 ± 25.0 | 82.8 ± 8.6 | 79.7 ± 9.4 |
| Miniature PSC frequencies (Hz) | 3.2 ± 0.71 | 2.4 ± 0.63 | 2.9 ± 0.48 | 2.0 ± 0.37 | 2.0 ± 0.53 | 2.0 ± 0.30 |
| Miniature PSC amplitudes (pA) | 22.6 ± 2.2 | 21.0 ± 3.1 | 21.9 ± 1.8 | 35.2 ± 2.9 | 20.0 ± 2.4 | 29.8 ± 2.4 * |
| Controls | ||||
| ID code | Gender | Age at biopsy | Previously published | |
| Control_1 | F | 48 | Japtok et al., 2015 [57] | |
| Control_2 | M | 34 | Glaß et al., 2018 [58] | |
| DYT-SGCE Patients | ||||
| ID code | Gender | Age at biopsy | Genotype of locus | Previously published |
| SGCE_1 | M | 39 | SGCE, c.298T>G | Grütz et al., 2017 [19] |
| SGCE_2 | F | 29 | SGCE, c.304C>T | Grütz et al., 2017 [19] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutschenko, A.; Staege, S.; Grütz, K.; Glaß, H.; Kalmbach, N.; Gschwendtberger, T.; Henkel, L.M.; Heine, J.; Grünewald, A.; Hermann, A.; et al. Functional and Molecular Properties of DYT-SGCE Myoclonus-Dystonia Patient-Derived Striatal Medium Spiny Neurons. Int. J. Mol. Sci. 2021, 22, 3565. https://doi.org/10.3390/ijms22073565
Kutschenko A, Staege S, Grütz K, Glaß H, Kalmbach N, Gschwendtberger T, Henkel LM, Heine J, Grünewald A, Hermann A, et al. Functional and Molecular Properties of DYT-SGCE Myoclonus-Dystonia Patient-Derived Striatal Medium Spiny Neurons. International Journal of Molecular Sciences. 2021; 22(7):3565. https://doi.org/10.3390/ijms22073565
Chicago/Turabian StyleKutschenko, Anna, Selma Staege, Karen Grütz, Hannes Glaß, Norman Kalmbach, Thomas Gschwendtberger, Lisa M. Henkel, Johanne Heine, Anne Grünewald, Andreas Hermann, and et al. 2021. "Functional and Molecular Properties of DYT-SGCE Myoclonus-Dystonia Patient-Derived Striatal Medium Spiny Neurons" International Journal of Molecular Sciences 22, no. 7: 3565. https://doi.org/10.3390/ijms22073565
APA StyleKutschenko, A., Staege, S., Grütz, K., Glaß, H., Kalmbach, N., Gschwendtberger, T., Henkel, L. M., Heine, J., Grünewald, A., Hermann, A., Seibler, P., & Wegner, F. (2021). Functional and Molecular Properties of DYT-SGCE Myoclonus-Dystonia Patient-Derived Striatal Medium Spiny Neurons. International Journal of Molecular Sciences, 22(7), 3565. https://doi.org/10.3390/ijms22073565