
The Significance of Targeting Poly (ADP-Ribose) Polymerase-1 in Pancreatic Cancer for Providing a New Therapeutic Paradigm

Abstract
1. Introduction
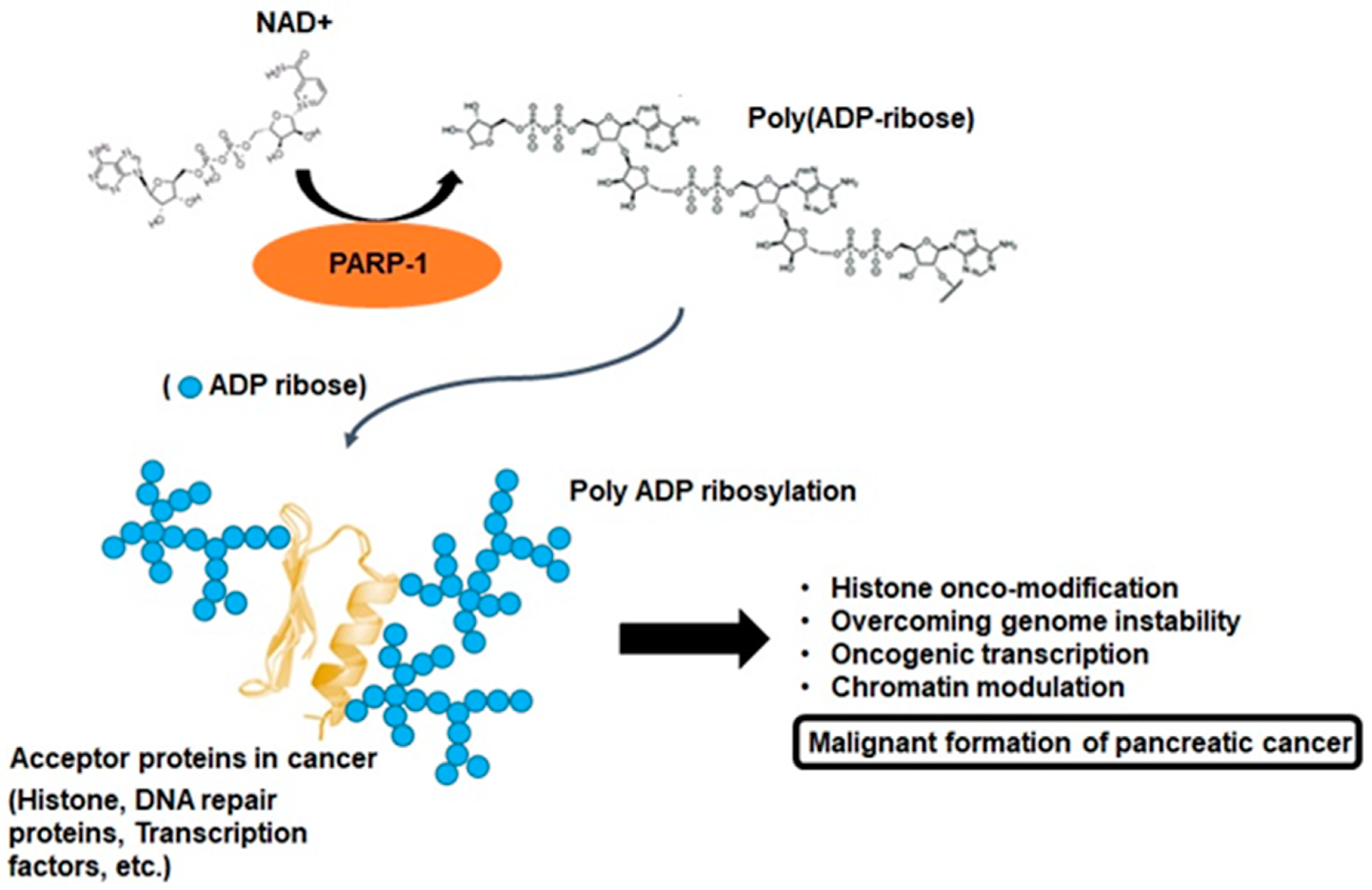
2. PARP-1 and Poly-ADP Ribosylation
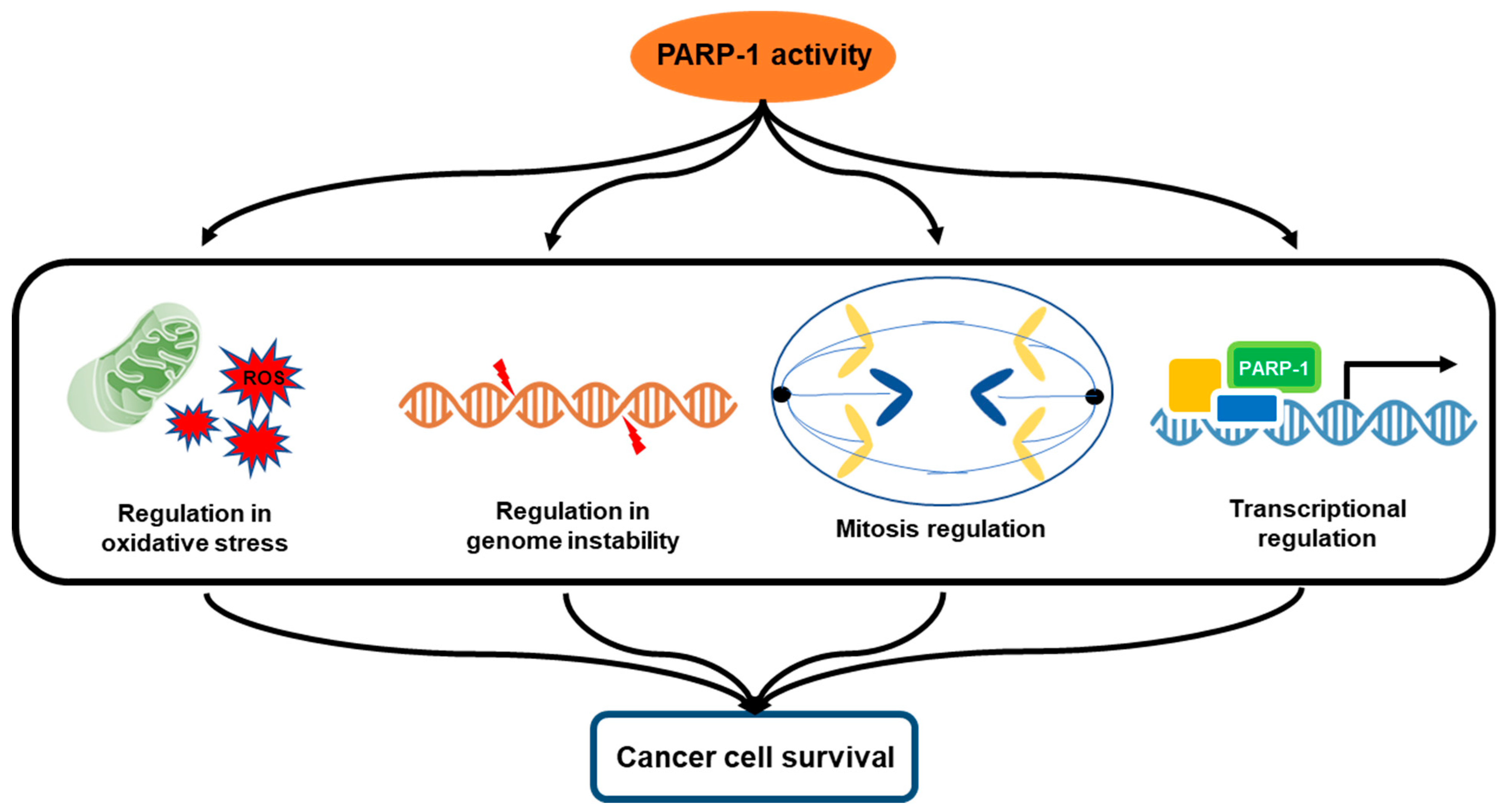
3. Multifactorial Functions of PARP-1 in Cancer Progression
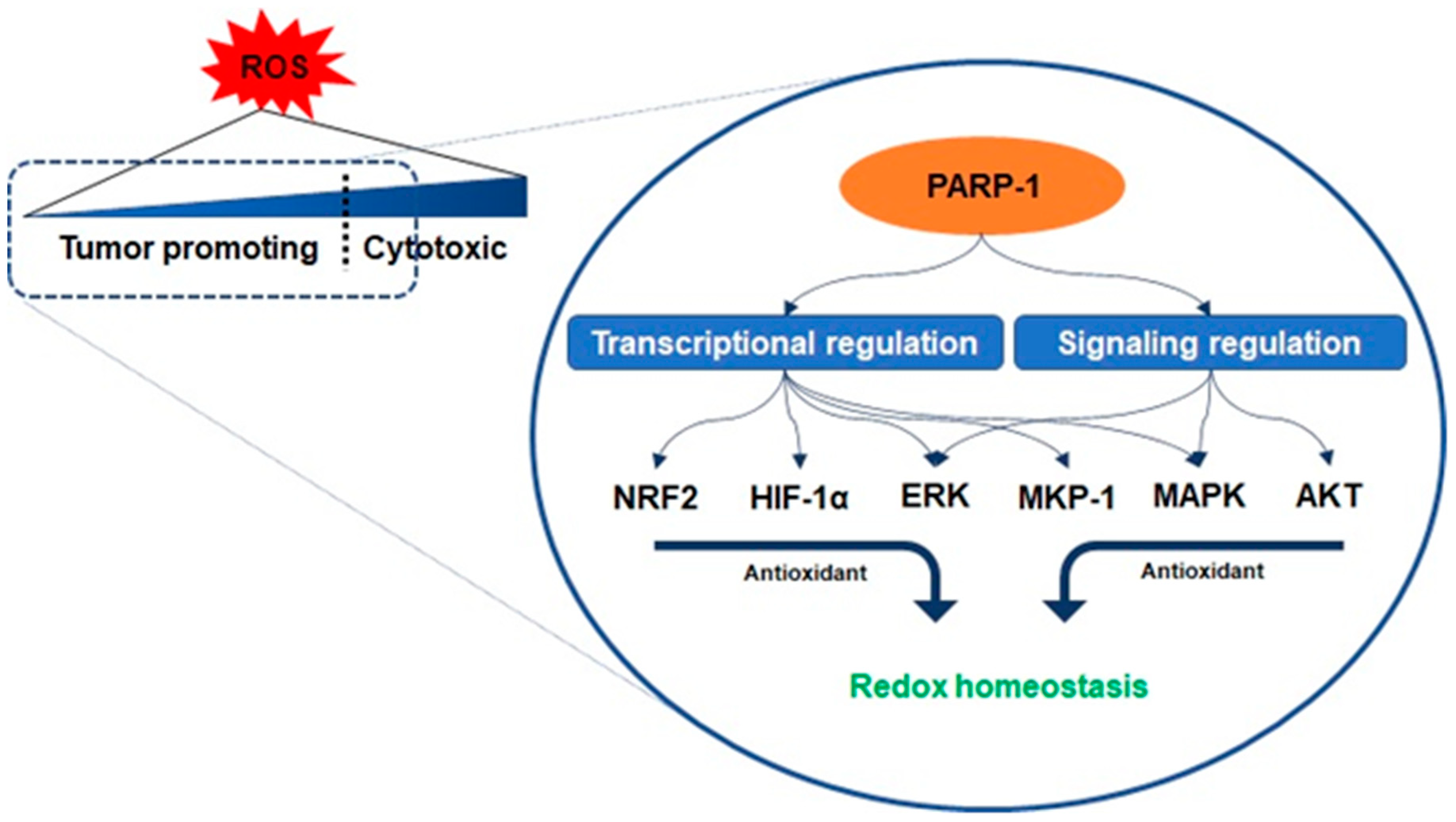
3.1. Oxidative Stress and PARP-1
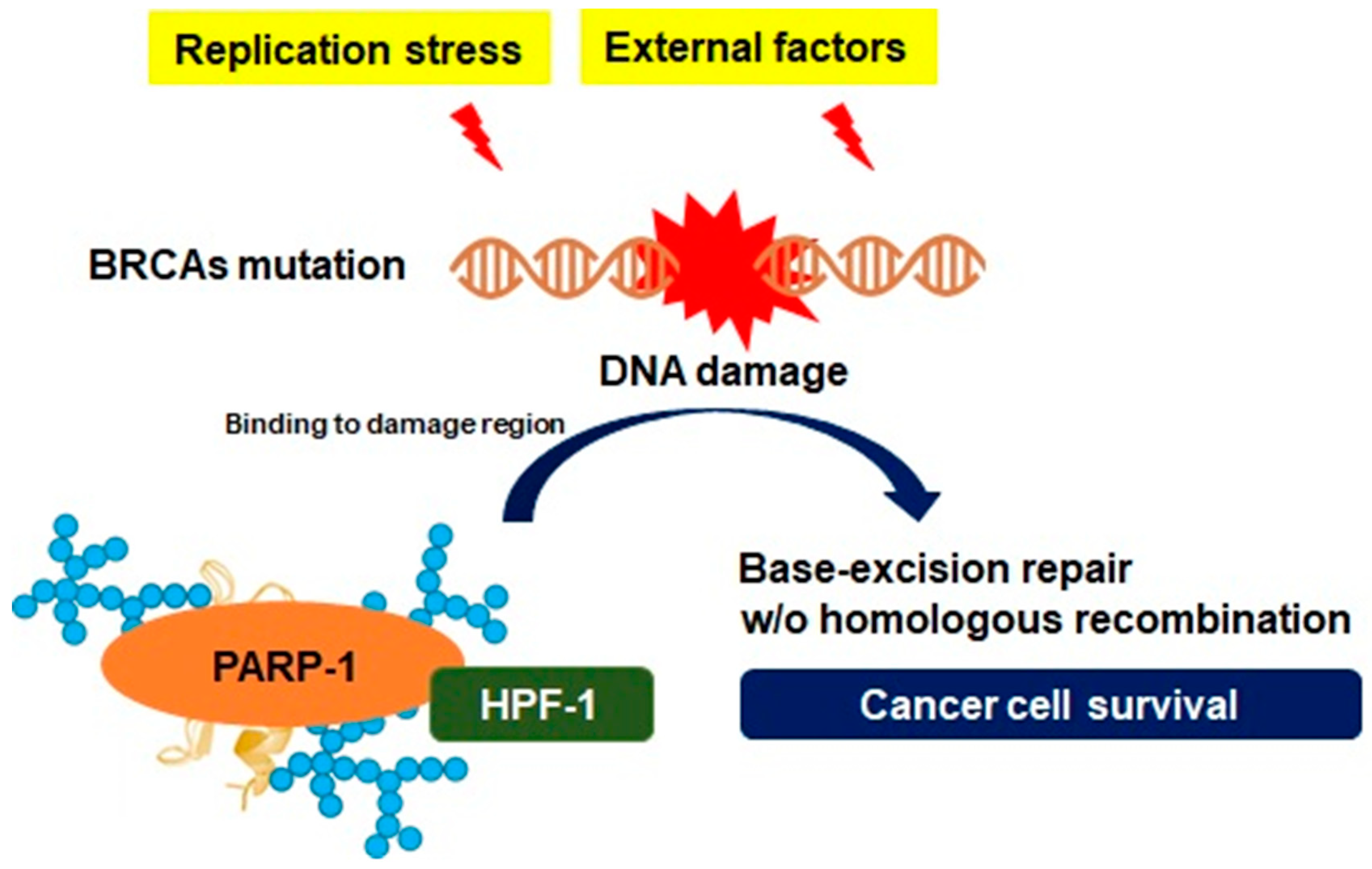
3.2. Genomic Instability and PARP-1
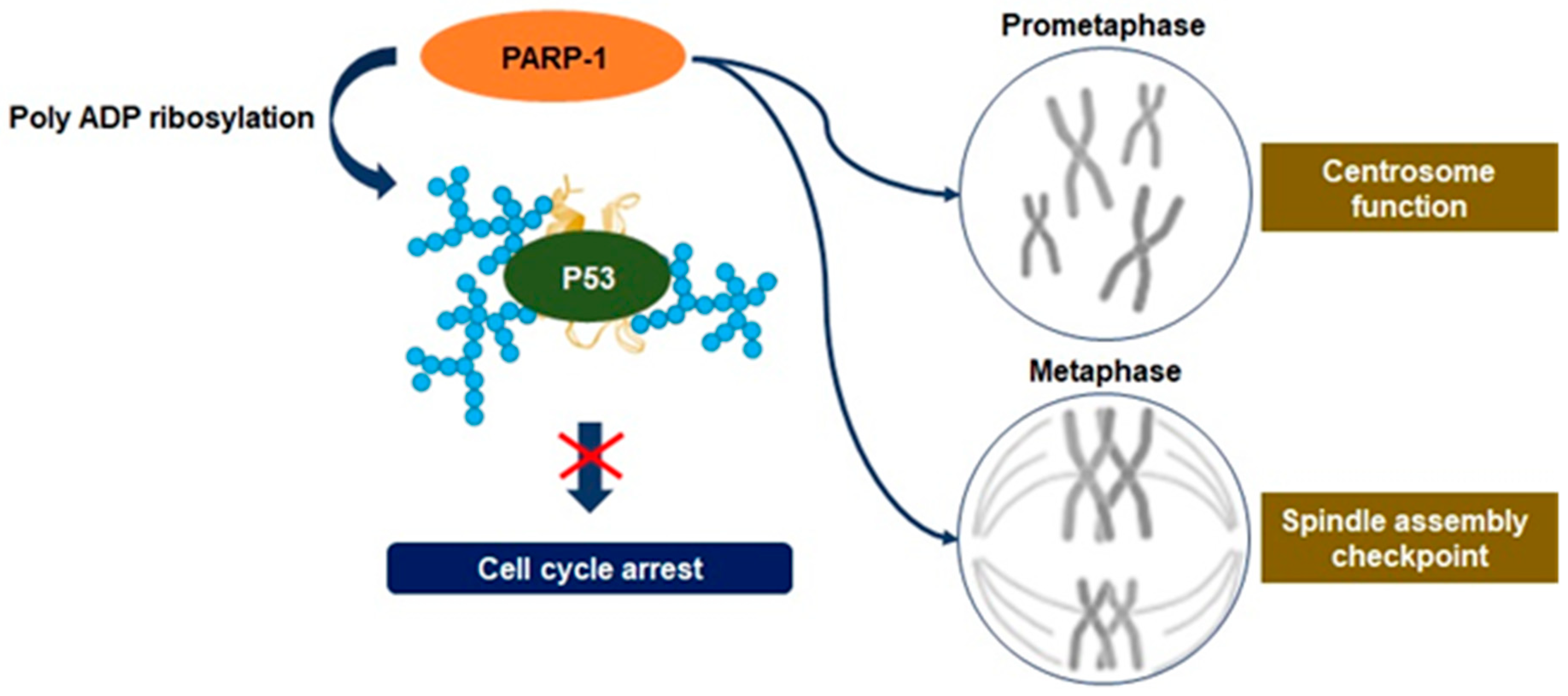
3.3. Mitosis Regulation
3.4. Transcriptional Regulation
4. Clinical Study: PARP-1 Inhibitor Application in Patients with Pancreatic Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FOLFIRINOX | Fluorouracil, leucovorin, irinotecan, and oxaliplatin |
| RAS | rat sarcoma viral oncogenes homolog |
| PARP-1 | Poly (adenosine diphosphate (ADP)-ribose) polymerase-1 |
| PARylation | Poly-ADP-ribosylation |
| PAR | poly-ADP-ribose |
| NAD+ | Nicotinamide adenine dinucleotide |
| ROS | Reactive oxygen species |
| NRF 2 | Nuclear factor erythroid-related factor 2 |
| HIF1-α | Hypoxia-inducing factor 1-alpha |
| AKT | Protein kinase B |
| ERK | Extracellular signal-regulated kinase |
| MKP-1 | Mitogen-activated protein kinase phosphatase 1 |
| MAPK | Mitogen-activated protein kinase |
| BRCA | Breast cancer susceptibility gene |
| ATM | Ataxia-telangiectasia mutated |
| ssDNA | Single-stranded DNA |
| dsDNA | Double-stranded DNA |
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Saung, M.T.; Zheng, L. Current Standards of Chemotherapy for Pancreatic Cancer. Clin. Ther. 2017, 39, 2125–2134. [Google Scholar] [CrossRef]
- Cho, I.R.; Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; An, C.; Park, M.S.; et al. FOLFIRINOX vs gemcitabine/nab-paclitaxel for treatment of metastatic pancreatic cancer: Single-center cohort study. World J. Gastrointest. Oncol. 2020, 12, 182–194. [Google Scholar] [CrossRef]
- Reni, M.; Balzano, G.; Zanon, S.; Passoni, P.; Nicoletti, R.; Arcidiacono, P.G.; Pepe, G.; Doglioni, C.; Fugazza, C.; Ceraulo, D.; et al. Phase 1B trial of Nab-paclitaxel plus gemcitabine, capecitabine, and cisplatin (PAXG regimen) in patients with unresectable or borderline resectable pancreatic adenocarcinoma. Br. J. Cancer 2016, 115, 290–296. [Google Scholar] [CrossRef][Green Version]
- Santini, D.; Virzi, V.; Vincenzi, B.; Rocci, L.; Leoni, V.; Tonini, G. A phase I trial of fixed dose rate gemcitabine plus capecitabine in metastatic cancer patients. Ann. Oncol. 2007, 18, 576–580. [Google Scholar] [CrossRef]
- Zong, Y.; Peng, Z.; Wang, X.; Lu, M.; Shen, L.; Zhou, J. Efficacy and Safety of Nab-Paclitaxel Plus S-1 versus Nab-Paclitaxel Plus Gemcitabine for First-Line Chemotherapy in Advanced Pancreatic Ductal Adenocarcinoma. Cancer Manag. Res. 2020, 12, 12657–12666. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Fang, B. RAS signaling and anti-RAS therapy: Lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim. Biophys. Sin. 2016, 48, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905. [Google Scholar] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar]
- Marti, J.M.; Fernandez-Cortes, M.; Serrano-Saenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar]
- Karlberg, T.; Thorsell, A.G.; Kallas, A.; Schuler, H. Crystal structure of human ADP-ribose transferase ARTD15/PARP16 reveals a novel putative regulatory domain. J. Biol. Chem. 2012, 287, 24077–24081. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys Acta 2011, 1807, 534–542. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid. Med. Cell Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, X.J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 2014, 67, 69–80. [Google Scholar] [PubMed]
- Pietrzak, J.; Spickett, C.M.; Ploszaj, T.; Virag, L.; Robaszkiewicz, A. PARP1 promoter links cell cycle progression with adaptation to oxidative environment. Redox. Biol. 2018, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Romero, R.; Martinez-Lara, E.; Aguilar-Quesada, R.; Peralta, A.; Oliver, F.J.; Siles, E. PARP-1 modulates deferoxamine-induced HIF-1alpha accumulation through the regulation of nitric oxide and oxidative stress. J. Cell Biochem. 2008, 104, 2248–2260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560. [Google Scholar]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar]
- Ethier, C.; Tardif, M.; Arul, L.; Poirier, G.G. PARP-1 modulation of mTOR signaling in response to a DNA alkylating agent. PLoS ONE 2012, 7, e47978. [Google Scholar] [CrossRef] [PubMed]
- Racz, B.; Hanto, K.; Tapodi, A.; Solti, I.; Kalman, N.; Jakus, P.; Kovacs, K.; Debreceni, B.; Gallyas, F., Jr.; Sumegi, B. Regulation of MKP-1 expression and MAPK activation by PARP-1 in oxidative stress: A new mechanism for the cytoplasmic effect of PARP-1 activation. Free Radic. Biol. Med. 2010, 49, 1978–1988. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar]
- Perkhofer, L.; Gout, J.; Roger, E.; Kude de Almeida, F.; Baptista Simoes, C.; Wiesmuller, L.; Seufferlein, T.; Kleger, A. DNA damage repair as a target in pancreatic cancer: State-of-the-art and future perspectives. Gut 2020, 70, 606–617. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6. [Google Scholar] [CrossRef]
- Smith, A.P.; Gimenez-Abian, J.F.; Clarke, D.J. DNA-damage-independent checkpoints: Yeast and higher eukaryotes. Cell Cycle 2002, 1, 16–33. [Google Scholar] [CrossRef][Green Version]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system-implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef]
- Ganem, N.J.; Pellman, D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012, 199, 871–881. [Google Scholar] [CrossRef]
- Rhind, N.; Russell, P. Signaling pathways that regulate cell division. Cold Spring Harb. Perspect. Biol. 2012, 4, a005942. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019, 167, 33–43. [Google Scholar] [CrossRef]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: A link between senescence and tumor immunity. Signal. Transduct. Target. Ther. 2020, 5, 107. [Google Scholar] [CrossRef]
- Kanai, M.; Tong, W.M.; Sugihara, E.; Wang, Z.Q.; Fukasawa, K.; Miwa, M. Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol. Cell Biol. 2003, 23, 2451–2462. [Google Scholar] [CrossRef]
- Kukolj, E.; Kaufmann, T.; Dick, A.E.; Zeillinger, R.; Gerlich, D.W.; Slade, D. PARP inhibition causes premature loss of cohesion in cancer cells. Oncotarget 2017, 8, 103931–103951. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, L.; Della Monica, R.; Visconti, R.; Costanzo, V.; Grieco, D. ATM controls proper mitotic spindle structure. Cell Cycle 2014, 13, 1091–1100. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Astanina, E.; Bussolino, F.; Doronzo, G. Multifaceted activities of transcription factor EB in cancer onset and progression. Mol. Oncol. 2021, 15, 327–346. [Google Scholar] [CrossRef]
- Annicotte, J.S.; Fayard, E.; Swift, G.H.; Selander, L.; Edlund, H.; Tanaka, T.; Kodama, T.; Schoonjans, K.; Auwerx, J. Pancreatic-duodenal homeobox 1 regulates expression of liver receptor homolog 1 during pancreas development. Mol. Cell Biol. 2003, 23, 6713–6724. [Google Scholar] [CrossRef]
- Nadolny, C.; Dong, X. Liver receptor homolog-1 (LRH-1): A potential therapeutic target for cancer. Cancer Biol. Ther. 2015, 16, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, Y. Transcriptional regulation of pancreas development and beta-cell function [Review]. Endocr. J. 2017, 64, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Vihervaara, A.; Duarte, F.M.; Lis, J.T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 2018, 19, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Wang, P.Y.; Wang, Y.T.; Yang, G.F.; Zhang, A.; Miao, Z.H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Zaremba, T.; Curtin, N.J. PARP inhibitor development for systemic cancer targeting. Anticancer Agents Med. Chem. 2007, 7, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Sandhu, S.K.; de Bono, J.S. PARP inhibitors: Mechanism of action and their potential role in the prevention and treatment of cancer. Drugs 2012, 72, 1579–1590. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Mladenov, E.; Anachkova, B.; Tsaneva, I. Sub-nuclear localization of Rad51 in response to DNA damage. Genes Cells 2006, 11, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmana, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Hua, J.; Liu, J.; Liang, C.; Meng, Q.; Ni, Q.; Shi, S.; Yu, X. Nab-paclitaxel plus gemcitabine as first-line treatment for advanced pancreatic cancer: A systematic review and meta-analysis. J. Cancer 2019, 10, 4420–4429. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Niraparib in Metastatic Pancreatic Cancer After Previous Chemotherapy (NIRA-PANC): A Phase 2 Trial. Available online: http://ClinicalTrials.gov/show/NCT03553004 (accessed on 15 March 2021).
- Kasi, A.; Chalise, P.; Williamson, S.K.; Baranda, J.C.; Sun, W.; Al-Rajabi, R.M.d.T.; Saeed, A.; Kumer, S.; Schmitt, T.; Foster, C.; et al. Niraparib in metastatic pancreatic cancer after previous chemotherapy (NIRA-PANC): A phase 2 trial. J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS4168. [Google Scholar] [CrossRef]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A phase 1 study of veliparib, a PARP-1/2 inhibitor, with gemcitabine and radiotherapy in locally advanced pancreatic cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- De Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmana, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A., 3rd; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Trial ID | Outcomes | Toxicity |
|---|---|---|---|
| Olaparib | NCT02677038 NCT02511223 | Objective response rate (PR: 6%; SD: 34%) | not available |
| Olaparib | NCT01078662 | Tumor response rate (CR: 2%; PR: 32%; SD: 23%) | Above grade 3: nausea, fatigue, vomiting, anemia, abdominal pain, diarrhea, dyspnea |
| Veliparib | NCT00892736 | Disease control rate (CR + PR: 23%; CR + PR + SD: 58%) | All grade: nausea, fatigue, and lymphopenia |
| Veliparib Gemcitabine | NCT01908478 | Median OS (15 moths); tumor response rate (CR: 3%; SD: 93%) | Above grade 3: lymphopenia and anemia |
| Rucaparib | NCT03140670 | Disease control rate (CR + PR + SD: 89.5%) | All grade: nausea, dysgeusia, fatigue |
| Rucaparib | NCT02042378 | Disease control rate (PR + SD: 32%) | Above grade 3: nausea, anemia |
| Talazoparib | NCT01286987 | Tumor response rate (PR: 15%; SD: 15%), median progression-free survival: 5.3 weeks | Above grade 3: fatigue, anemia, neutropenia, thrombocytopenia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, K.-Y.; Park, M.H. The Significance of Targeting Poly (ADP-Ribose) Polymerase-1 in Pancreatic Cancer for Providing a New Therapeutic Paradigm. Int. J. Mol. Sci. 2021, 22, 3509. https://doi.org/10.3390/ijms22073509
Jeong K-Y, Park MH. The Significance of Targeting Poly (ADP-Ribose) Polymerase-1 in Pancreatic Cancer for Providing a New Therapeutic Paradigm. International Journal of Molecular Sciences. 2021; 22(7):3509. https://doi.org/10.3390/ijms22073509
Chicago/Turabian StyleJeong, Keun-Yeong, and Min Hee Park. 2021. "The Significance of Targeting Poly (ADP-Ribose) Polymerase-1 in Pancreatic Cancer for Providing a New Therapeutic Paradigm" International Journal of Molecular Sciences 22, no. 7: 3509. https://doi.org/10.3390/ijms22073509
APA StyleJeong, K.-Y., & Park, M. H. (2021). The Significance of Targeting Poly (ADP-Ribose) Polymerase-1 in Pancreatic Cancer for Providing a New Therapeutic Paradigm. International Journal of Molecular Sciences, 22(7), 3509. https://doi.org/10.3390/ijms22073509