
The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
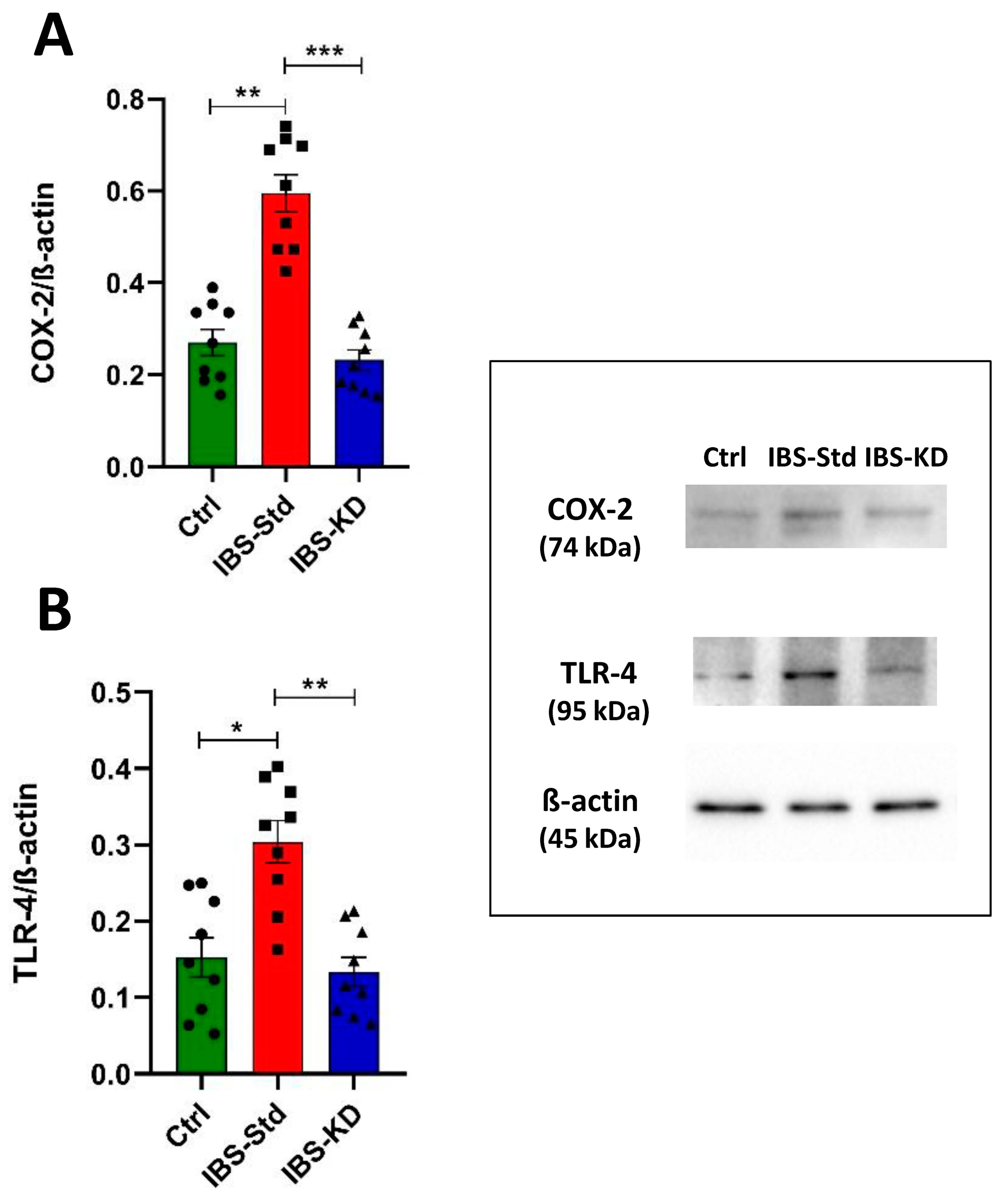
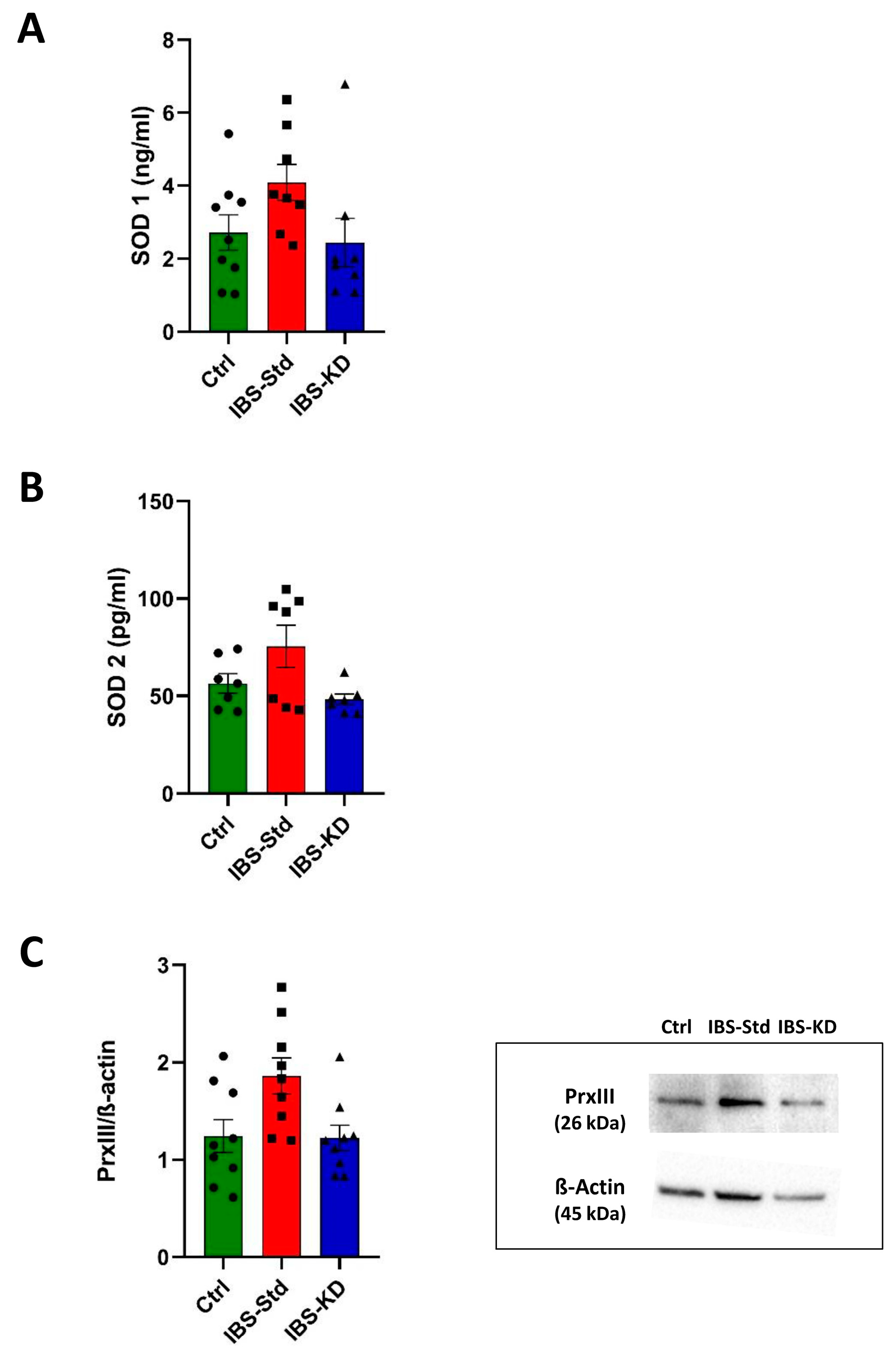
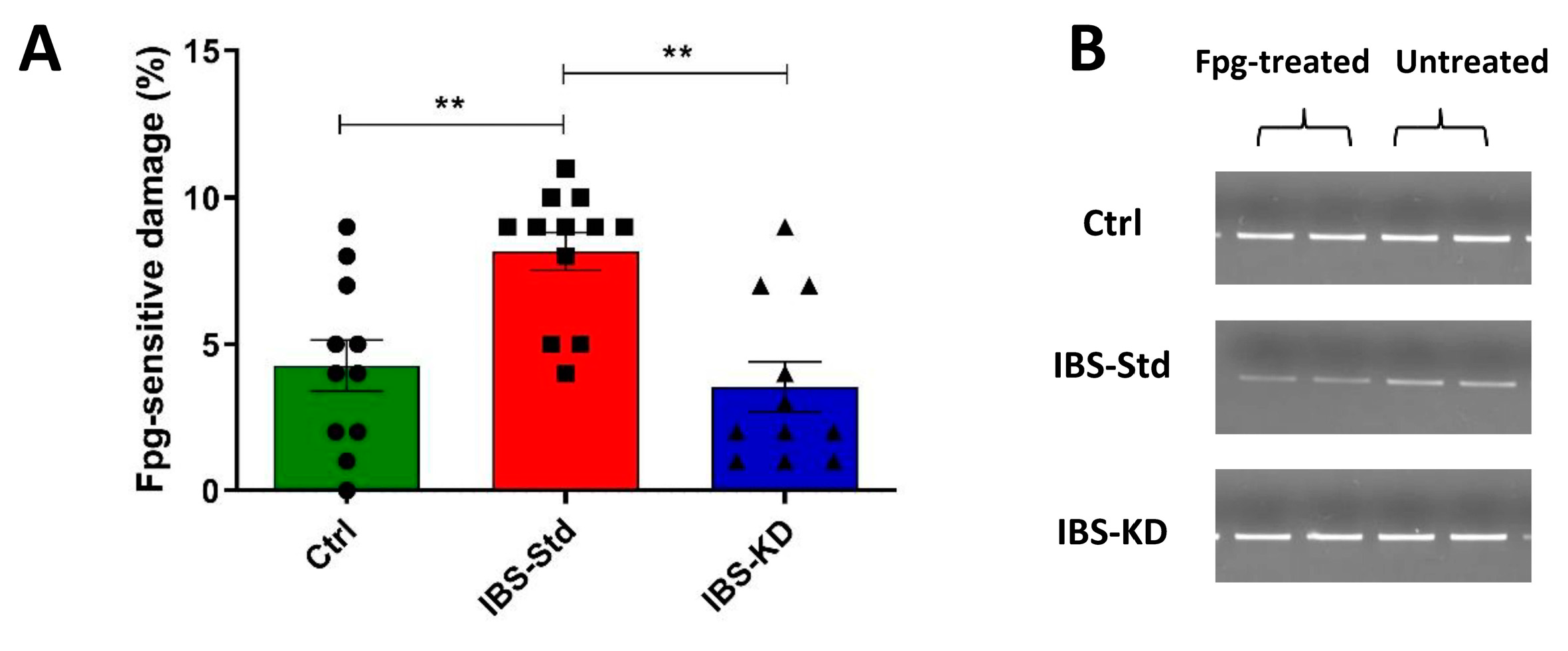
2.1. Evaluation of Markers of Inflammation and Determination of the Redox Status
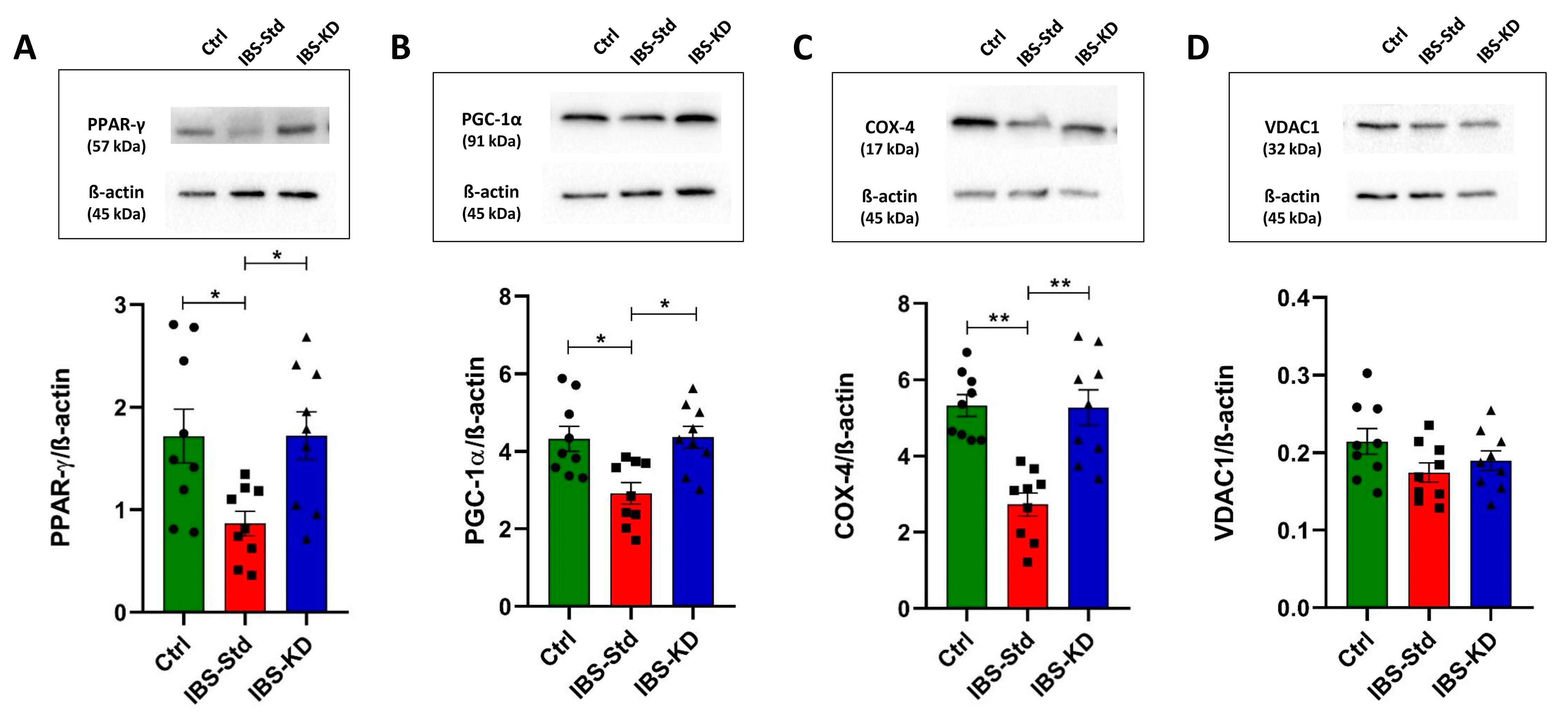
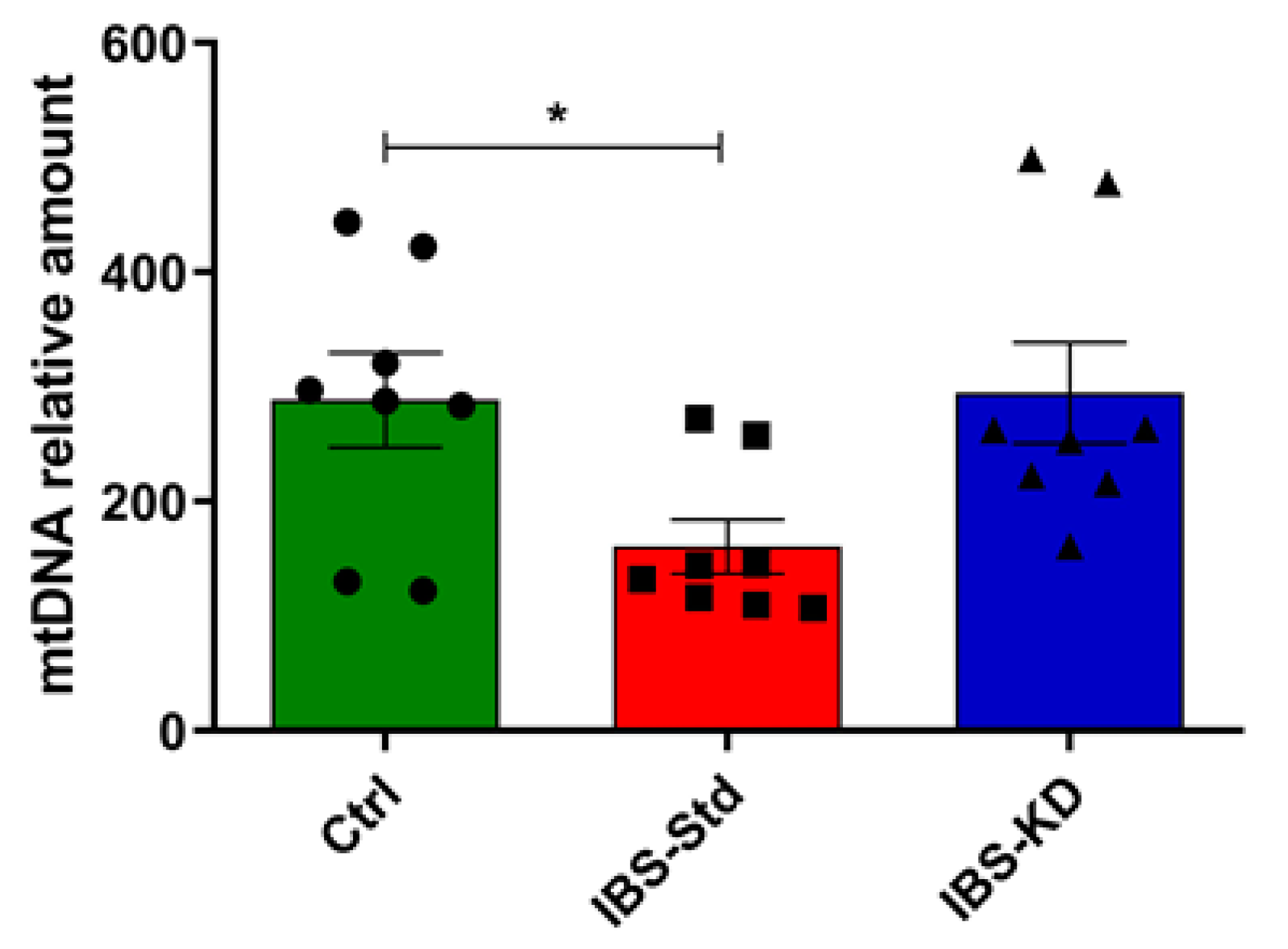
2.2. Mitochondrial Biogenesis
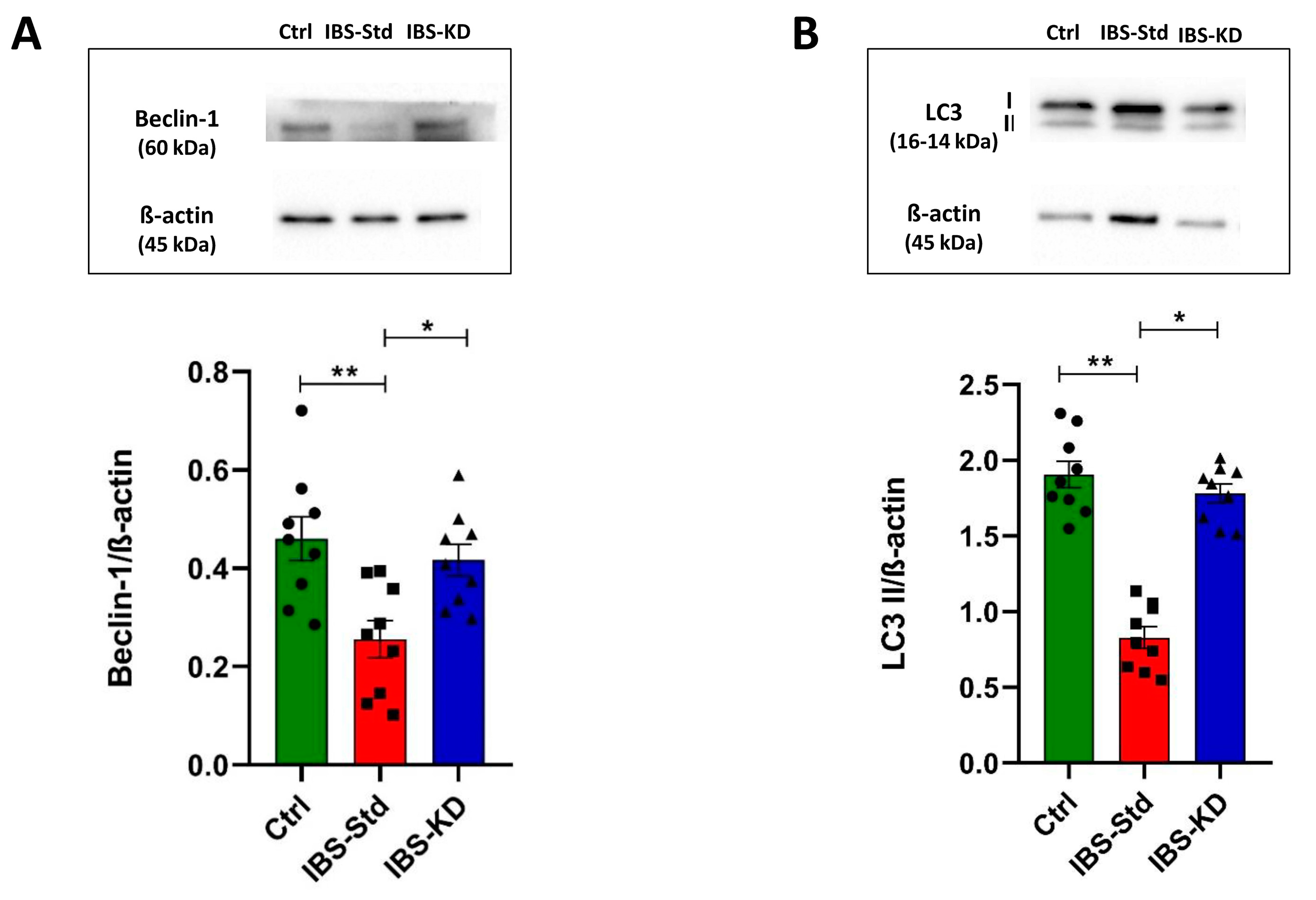
2.3. Evaluation of Markers of Autophagy
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Western Immunoblotting
4.3. SOD 1 and SOD 2 Levels
4.4. Determination of mtDNA Content
4.5. Modified Purines Analysis
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy, R.L.; Olden, K.W.; Naliboff, B.D.; Bradley, L.A.; Francisconi, C.; Drossman, D.A.; Creed, F. Psychosocial Aspects of the Functional Gastrointestinal Disorders. Gastroenterology 2006, 130, 1447–1458. [Google Scholar] [CrossRef]
- Labanski, A.; Langhorst, J.; Engler, H.; Elsenbruch, S. Stress and the brain-gut axis in functional and chronic-inflammatory gastrointestinal diseases: A transdisciplinary challenge. Psychoneuroendocrinology 2020, 111, 104501. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Gupta, M.S. The IBS market. Nat. Rev. Drug Discov. 2006, 5, 99–100. [Google Scholar] [CrossRef]
- Russo, F.; Chimienti, G.; Clemente, C.; D’Attoma, B.; Linsalata, M.; Orlando, A.; De Carne, M.; Cariola, F.; Semeraro, F.P.; Pepe, G.; et al. Adipokine profile in celiac patients: Differences in comparison with patients suffering from diarrhea-predominant IBS and healthy subjects. Scand. J. Gastroenterol. 2013, 48, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, D. Immunomodulation of enteric neural function in irritable bowel syndrome. World J. Gastroenterol. 2015, 21, 7362–7366. [Google Scholar] [CrossRef] [PubMed]
- Karabatsiakis, A.; Schonfeldt-Lecuona, C. Depression, mitochondrial bioenergetics, and electroconvulsive therapy: A new approach towards personalized medicine in psychiatric treatment—A short review and current perspective. Transl. Psychiatry 2020, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Orlando, A.; Chimienti, G.; Pesce, V.; Fracasso, F.; Lezza, A.M.S.; Russo, F. An In Vitro Study on Mitochondrial Compensatory Response Induced by Gliadin Peptides in Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 1862. [Google Scholar] [CrossRef]
- Picca, A.; Riezzo, G.; Clemente, C.; Pesce, V.; Orlando, A.; Chimienti, G.; Russo, F.; Lezza, A.M.S. Mitochondria and redox balance in coeliac disease: A case-control study. Eur. J. Clin. Investig. 2018, 48, e12877. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef]
- Vicario, M.; Alonso, C.; Guilarte, M.; Serra, J.; Martínez, C.; González-Castro, A.M.; Lobo, B.; Antolín, M.; Andreu, A.L.; García-Arumí, E.; et al. Chronic psychosocial stress induces reversible mitochondrial damage and corticotropin-releasing factor receptor type-1 upregulation in the rat intestine and IBS-like gut dysfunction. Psychoneuroendocrinology 2012, 37, 65–77. [Google Scholar] [CrossRef]
- Mottawea, W.; Chiang, C.-K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Sugiyama, T.; Shiotani, A. The Cutting Edge Research of Functional Gastrointestinal Disorders in Japan: Review on JGA Core Symposium 2018–2020. Digestion 2021, 102, 6–11. [Google Scholar] [CrossRef]
- Austin, G.L.; Dalton, C.B.; Hu, Y.; Morris, C.B.; Hankins, J.; Weinland, S.R.; Westman, E.C.; Yancy, W.S.; Drossman, D.A. A Very Low-Carbohydrate Diet Improves Symptoms and Quality of Life in Diarrhea-Predominant Irritable Bowel Syndrome. Clin. Gastroenterol. Hepatol. 2009, 7, 706–708.e1. [Google Scholar] [CrossRef]
- Locker, F.; Leitner, J.; Aminzadeh-Gohari, S.; Weber, D.D.; Sanio, P.; Koller, A.; Feichtinger, R.G.; Weiss, R.; Kofler, B.; Lang, R. The Influence of Ketogenic Diets on Psoriasiform-Like Skin Inflammation. J. Investig. Dermatol. 2020, 140, 707–710.e7. [Google Scholar] [CrossRef] [PubMed]
- Rawat, K.; Singh, N.; Kumari, P.; Saha, L. A review on preventive role of ketogenic diet (KD) in CNS disorders from the gut microbiota perspective. Rev. Neurosci. 2021, 32, 143–157. [Google Scholar] [CrossRef]
- Brietzke, E.; Mansur, R.B.; Subramaniapillai, M.; Balanzá-Martínez, V.; Vinberg, M.; González-Pinto, A.; Rosenblat, J.D.; Ho, R.; McIntyre, R.S. Ketogenic diet as a metabolic therapy for mood disorders: Evidence and developments. Neurosci. Biobehav. Rev. 2018, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Storoni, M.; Robert, M.P.; Plant, G.T. The therapeutic potential of a calorie-restricted ketogenic diet for the management of Leber hereditary optic neuropathy. Nutr. Neurosci. 2017, 22, 156–164. [Google Scholar] [CrossRef]
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.S.; Khan, A.; Rho, J.M.; Shearer, J. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBR(T+tf/j) Mouse. Front. Physiol. 2016, 7, 654. [Google Scholar] [CrossRef] [PubMed]
- Khorjahani, A.; Peeri, M.; Azarbayjani, M.A. Therapeutic Effect of Exercise on Anxiety and Bowel Oxidative Stress in the Maternal Separation Animal Model. Basic Clin. Neurosci. J. 2019, 11, 69–78. [Google Scholar] [CrossRef]
- Ogino, S.; Kirkner, G.J.; Nosho, K.; Irahara, N.; Kure, S.; Shima, K.; Hazra, A.; Chan, A.T.; Dehari, R.; Giovannucci, E.L.; et al. Cyclooxygenase-2 Expression Is an Independent Predictor of Poor Prognosis in Colon Cancer. Clin. Cancer Res. 2008, 14, 8221–8227. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Guo, Y.; Feng, X.J.; Liu, J.J.; Chang, Z.P.; Deng, G.F.; Xu, D.; Gao, J.P.; Hou, R.G. Oridonin Attenuates TNBS-induced Post-inflammatory Irritable Bowel Syndrome via PXR/NF-kappaB Signaling. Inflammation 2020, 44, 645–658. [Google Scholar] [CrossRef]
- Russo, F.; Chimienti, G.; Riezzo, G.; Linsalata, M.; D’Attoma, B.; Clemente, C.; Orlando, A. Adipose Tissue-Derived Biomarkers of Intestinal Barrier Functions for the Characterization of Diarrhoea-Predominant IBS. Dis. Markers 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef]
- Gigante, I.; Tutino, V.; Russo, F.; De Nunzio, V.; Coletta, S.; Armentano, R.; Crovace, A.; Caruso, M.; Orlando, A.; Notarnicola, M. Cannabinoid Receptors Overexpression in a Rat Model of Irritable Bowel Syndrome (IBS) after Treatment with a Ketogenic Diet. Int. J. Mol. Sci. 2021, 22, 2880. [Google Scholar] [CrossRef]
- Balmus, I.-M.; Ilie, O.-D.; Ciobica, A.; Cojocariu, R.; Stanciu, C.; Trifan, A.; Cimpeanu, M.; Cimpeanu, C.; Gorgan, D.L. Irritable Bowel Syndrome between Molecular Approach and Clinical Expertise—Searching for Gap Fillers in the Oxidative Stress Way of Thinking. Medicina 2020, 56, 38. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maneix, L.; Catic, A. Touch and go: Nuclear proteolysis in the regulation of metabolic genes and cancer. FEBS Lett. 2016, 590, 908–923. [Google Scholar] [CrossRef]
- Vetuschi, A.; Pompili, S.; Gaudio, E.; Latella, G.; Sferra, R. PPAR-gamma with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8839–8848. [Google Scholar] [PubMed]
- Pedersen, G. Development, validation and implementation of an in vitro model for the study of metabolic and immune function in normal and inflamed human colonic epithelium. Dan. Med. J. 2015, 62, 4973. [Google Scholar]
- Roudsari, N.M.; Lashgari, N.A.; Zandi, N.; Pazoki, B.; Momtaz, S.; Sahebkar, A.; Abdolghaffari, A.H. PPAR gamma: A turning point for irritable bowel syndrome treatment. Life Sci. 2020, 257, 118103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, W.; Li, G.; Chen, J.; Guan, X.; Chen, X.; Guan, Z. Neuroprotective Effect and Mechanism of Thiazolidinedione on Dopaminergic Neurons In Vivo and In Vitro in Parkinson’s Disease. PPAR Res. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, S.B.; Bhat, A.; Ray, B.; Sugumar, M.; Muthukumar, S.P.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Guillemin, G.J.; Sakharkar, M.K. Cocoa beans improve mitochondrial biogenesis via PPARgamma/PGC1alpha dependent signalling pathway in MPP(+) intoxicated human neuroblastoma cells (SH-SY5Y). Nutr. Neurosci. 2020, 23, 471–480. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Cunningham, K.E.; Vincent, G.; Sodhi, C.P.; Novak, E.A.; Ranganathan, S.; Egan, C.E.; Stolz, D.B.; Rogers, M.B.; Firek, B.; Morowitz, M.J.; et al. Peroxisome Proliferator-activated Receptor-gamma Coactivator 1-alpha (PGC1alpha) Protects against Experimental Murine Colitis. J. Biol. Chem. 2016, 291, 10184–10200. [Google Scholar] [CrossRef]
- Halling, J.F.; Ringholm, S.; Nielsen, M.M.; Overby, P.; Pilegaard, H. PGC-1α promotes exercise-induced autophagy in mouse skeletal muscle. Physiol. Rep. 2016, 4, e12698. [Google Scholar] [CrossRef]
- Menzies, R.A.; Gold, P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 1971, 246, 2425–2429. [Google Scholar] [CrossRef]
- Wang, T.; Liu, K.; Wen, L.; Yang, Y.; Yin, X.; Liu, K.; Chen, Y.; He, Y.; Yang, M.; Wei, Y.; et al. Autophagy and Gastrointestinal Diseases. Adv. Exp. Med. Biol. 2020, 1207, 529–556. [Google Scholar]
- Wang, S.-L.; Shao, B.-Z.; Zhao, S.-B.; Chang, X.; Wang, P.; Miao, C.-Y.; Li, Z.-S.; Bai, Y. Intestinal autophagy links psychosocial stress with gut microbiota to promote inflammatory bowel disease. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Vincent, G.; Novak, E.A.; Siow, V.S.; Cunningham, K.E.; Griffith, B.D.; Iv, T.E.C.; Mentrup, H.L.; Stolz, D.B.; Loughran, P.; Ranganathan, S.; et al. Nix-Mediated Mitophagy Modulates Mitochondrial Damage During Intestinal Inflammation. Antioxidants Redox Signal. 2020, 33, 1–19. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th edition). Autophagy 2021, 8, 1–382. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 1–10. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275.e16. [Google Scholar] [CrossRef]
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The ketogenic diet—A metabolism-based therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59. [Google Scholar] [CrossRef]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 1–27. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family?from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflügers Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Hasan-Olive, M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2018, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Hutfles, L.J.; Wilkins, H.M.; Koppel, S.J.; Weidling, I.W.; Selfridge, J.E.; Tan, E.; Thyfault, J.P.; Slawson, C.; Fenton, A.W.; Zhu, H.; et al. A bioenergetics systems evaluation of ketogenic diet liver effects. Appl. Physiol. Nutr. Metab. 2017, 42, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Cunningham, R.P.; Kelty, T.J.; Boccardi, L.R.; Nguyen, N.; Booth, F.W.; Rector, R.S. Ketogenic diet in combination with voluntary exercise impacts markers of hepatic metabolism and oxidative stress in male and female Wistar rats. Appl. Physiol. Nutr. Metab. 2020, 45, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Parry, H.A.; Kephart, W.C.; Mumford, P.W.; Romero, M.A.; Mobley, C.B.; Zhang, Y.; Roberts, M.D.; Kavazis, A.N. Ketogenic diet increases mitochondria volume in the liver and skeletal muscle without altering oxidative stress markers in rats. Heliyon 2018, 4, e00975. [Google Scholar] [CrossRef]
- McCommis, K.S.; Kovacs, A.; Weinheimer, C.J.; Shew, T.M.; Koves, T.R.; Ilkayeva, O.R.; Kamm, D.R.; Pyles, K.D.; King, M.T.; Veech, R.L.; et al. Nutritional modulation of heart failure in mitochondrial pyruvate carrier–deficient mice. Nat. Metab. 2020, 2, 1232–1247. [Google Scholar] [CrossRef]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef]
- Duszka, K.; Gregor, A.; Guillou, H.; König, J.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Caloric Restriction—Common Pathways Affecting Metabolism, Health, and Longevity. Cells 2020, 9, 1708. [Google Scholar] [CrossRef] [PubMed]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Up-regulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Whiting, S.; Andrew, R.D.; Macdonald, E.A.; Musa-Veloso, K.; Cunnane, S.C. Elevated polyunsaturated fatty acids in blood serum obtained from children on the ketogenic diet. Neurology 2003, 60, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A. Ketogenic Diet and PPAR Gamma. In Ketogenic Diet and Metabolic Therapies: Expanded Roles in Health and Disease; Masino, S.A., Ed.; Oxford University Press: New York, NY, USA, 2017; pp. 167–185. [Google Scholar]
- Vannucchi, M.G.; Evangelista, S. Experimental Models of Irritable Bowel Syndrome and the Role of the Enteric Neurotransmission. J. Clin. Med. 2018, 7, 4. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Rats (Number) | Maternal Deprivation (3 h/Day from PNDs 2 to 14) | Treatment (for 10 Weeks after PND 14) |
|---|---|---|---|
| Ctrl | 12 | No | Standard diet |
| IBS-Std | 11 | Yes | Standard diet |
| IBS-KD | 17 | Yes | Ketogenic diet |
| Analytical Constituents | Standard Diet (4RF21) | Ketogenic Diet |
|---|---|---|
| Moisture | 12% | 0% |
| Crude protein | 18.5% | 16.0 |
| Crude oils and fats | 3.0% | 67.0 |
| Crude fibers | 6.0% | 6.0 |
| Crude ash | 7.0% | 4.5 |
| Nutritional additives | ||
| Vitamin A | 28,500 I.U. | 25,770 I.U. |
| Vitamin D3 | 1260 I.U. | 2860 I.U. |
| Fe | 180 mg | 60 mg |
| Mn | 54 mg | 87.5 mg |
| Zn | 67.5 mg | 48.5 mg |
| Cu | 11.7 mg | 8.5 mg |
| I | 0.9 mg | 0.3 mg |
| Se | - | 0.16 mg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chimienti, G.; Orlando, A.; Lezza, A.M.S.; D’Attoma, B.; Notarnicola, M.; Gigante, I.; Pesce, V.; Russo, F. The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome. Int. J. Mol. Sci. 2021, 22, 3498. https://doi.org/10.3390/ijms22073498
Chimienti G, Orlando A, Lezza AMS, D’Attoma B, Notarnicola M, Gigante I, Pesce V, Russo F. The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome. International Journal of Molecular Sciences. 2021; 22(7):3498. https://doi.org/10.3390/ijms22073498
Chicago/Turabian StyleChimienti, Guglielmina, Antonella Orlando, Angela Maria Serena Lezza, Benedetta D’Attoma, Maria Notarnicola, Isabella Gigante, Vito Pesce, and Francesco Russo. 2021. "The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome" International Journal of Molecular Sciences 22, no. 7: 3498. https://doi.org/10.3390/ijms22073498
APA StyleChimienti, G., Orlando, A., Lezza, A. M. S., D’Attoma, B., Notarnicola, M., Gigante, I., Pesce, V., & Russo, F. (2021). The Ketogenic Diet Reduces the Harmful Effects of Stress on Gut Mitochondrial Biogenesis in a Rat Model of Irritable Bowel Syndrome. International Journal of Molecular Sciences, 22(7), 3498. https://doi.org/10.3390/ijms22073498