
Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation


Abstract
1. Introduction
2. Results
2.1. Inhibitory Effect of Evodiamine on the Growth of H. pylori by Downregulation of Replication and Transcription Genes
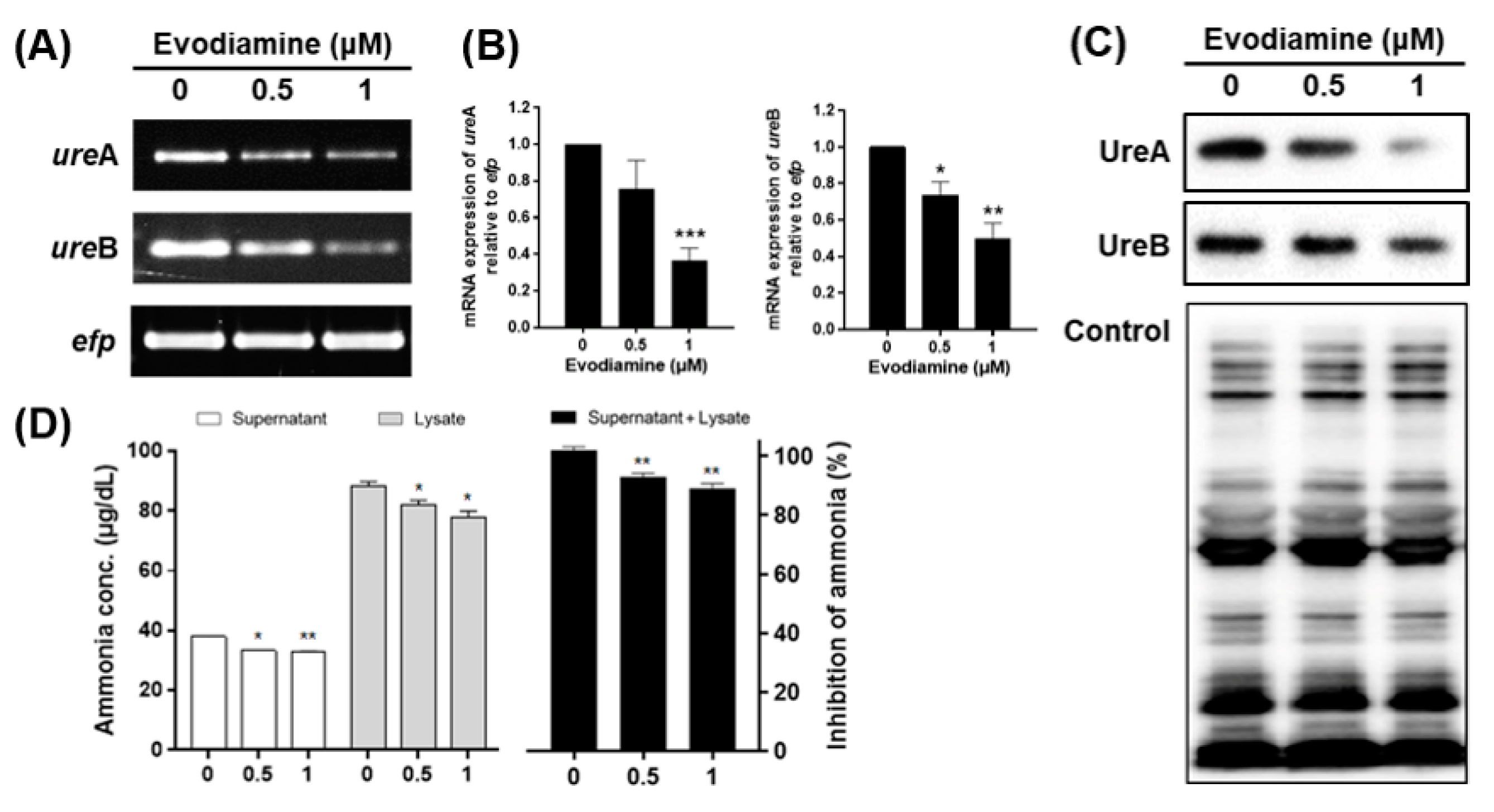
2.2. Downregulation of Urease in H. pylori Treated with Evodiamine
2.3. Reduced CagA and VacA Translocation to AGS Cells by Evodiamine
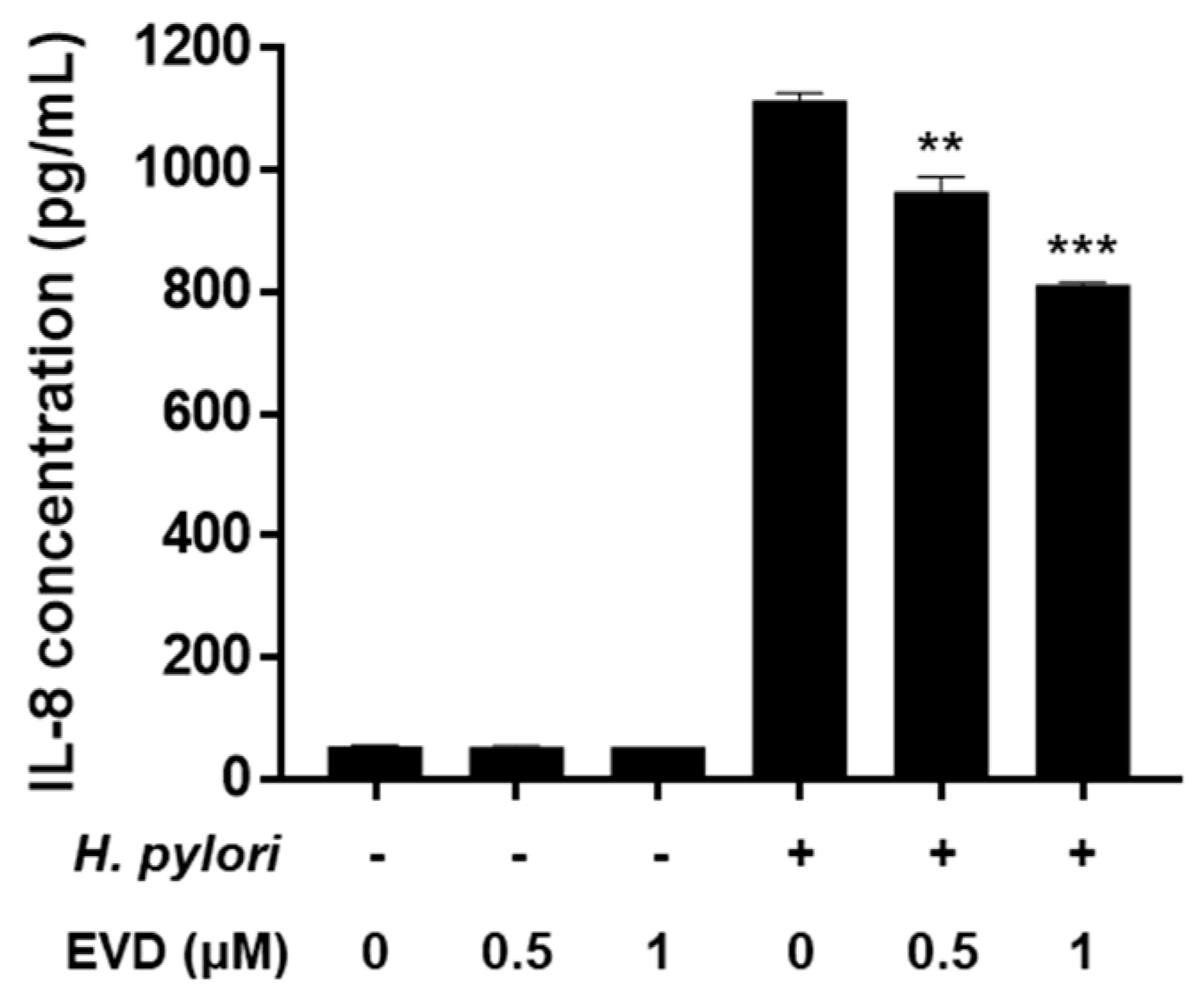
2.4. Decrease of Inflammatory Cytokines Induced by H. pylori Infection via Inhibition of MAPK and NF-κB Activation by Evodiamine
3. Discussion
4. Materials and Methods
4.1. Bacterial and Mammalian Cell Culture
4.2. Determination of MIC
4.3. RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
4.4. Protein Extraction and Western Blot
4.5. Urease Activity Test
4.6. Subcellular Fractionation
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zali, H.; Rezaei-Tavirani, M.; Azodi, M. Gastric cancer: Prevention, risk factors and treatment. Gastroenterol. Hepatol. Bed Bench 2011, 4, 175–185. [Google Scholar]
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric cancer: Epidemiology, prevention, classification, and treatment. Cancer Manag. Res. 2018, 10, 239–248. [Google Scholar] [CrossRef]
- Oakley, A.J. A structural view of bacterial DNA replication. Protein Sci. 2019, 28, 990–1004. [Google Scholar] [CrossRef]
- Terradot, L.; Zawilak-Pawlik, A. Structural insight into Helicobacter pylori DNA replication initiation. Gut Microbes 2010, 1, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Nitharwal, R.G.; Verma, V.; Dasgupta, S.; Dhar, S.K. Helicobacter pylori chromosomal DNA replication: Current status and future perspectives. FEBS Lett. 2010, 585, 7–17. [Google Scholar] [CrossRef][Green Version]
- Borin, B.N.; Tang, W.; Krezel, A.M. Helicobacter pylori RNA polymerase α-subunit C-terminal domain shows features unique to ε-proteobacteria and binds NikR/DNA complexes. Protein Sci. 2014, 23, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori Infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. 1), 57–64. [Google Scholar] [CrossRef] [PubMed]
- Blum, F.C.; Hu, H.Q.; Servetas, S.L.; Benoit, S.L.; Maier, R.J.; Maroney, M.J.; Merrell, D.S. Structure-function analyses of metal-binding sites of HypA reveal residues important for hydrogenase maturation in Helicobacter pylori. PLoS ONE 2017, 12, e0183260. [Google Scholar] [CrossRef] [PubMed]
- Voland, P.; Weeks, D.L.; Marcus, E.A.; Prinz, C.; Sachs, G.; Scott, D. Interactions among the seven Helicobacter pylori proteins encoded by the urease gene cluster. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G96–G106. [Google Scholar] [CrossRef]
- Ansari, S.; Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 2017, 22. [Google Scholar] [CrossRef]
- Futagami, S.; Hiratsuka, T.; Tatsuguchi, A.; Suzuki, K.; Kusunoki, M.; Shinji, Y.; Shinoki, K.; Iizumi, T.; Akamatsu, T.; Nishigaki, H.; et al. Monocyte chemoattractant protein 1 (MCP-1) released from Helicobacter pylori stimulated gastric epithelial cells induces cyclooxygenase 2 expression and activation in T cells. Gut 2003, 52, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Sebkova, L.; Pellicanò, A.; Monteleone, G.; Grazioli, B.; Guarnieri, G.; Imeneo, M.; Pallone, F.; Luzza, F. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect. Immun. 2004, 72, 5019–5026. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, K.-S.; Kim, J.-H.; Yang, S.-H.; Yoon, J.Y.; Han, B.-G.; Kim, H.S.; Lee, S.J.; Jang, J.Y.; Kim, K.H.; et al. Helicobacter pylori proinflammatory protein up-regulates NF-κB as a cell-translocating Ser/Thr kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 21418–21423. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef]
- Hu, B.; Khara, P.; Song, L.; Lin, A.S.; Frick-Cheng, A.E.; Harvey, M.L.; Cover, T.L.; Christie, P.J. In situ molecular architecture of the Helicobacter pylori cag type IV secretion system. mBio 2019, 10, e00849-19. [Google Scholar] [CrossRef]
- Chung, J.M.; Sheedlo, M.J.; Campbell, A.M.; Sawhney, N.; Frick-Cheng, A.E.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structure of the Helicobacter pylori Cag type IV secretion system. eLife 2019, 8. [Google Scholar] [CrossRef]
- Higuchi, M.; Tsutsumi, R.; Higashi, H.; Hatakeyama, M. Conditional gene silencing utilizing the lac repressor reveals a role of SHP-2 in cagA-positive Helicobacter pylori pathogenicity. Cancer Sci. 2004, 95, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Lim, J.W.; Kim, H. Helicobacter pylori in a Korean isolate activates mitogen-activated protein kinases, AP-1, and NF-κB and induces chemokine expression in gastric epithelial AGS cells. Lab. Investig. 2003, 84, 49–62. [Google Scholar] [CrossRef]
- Boquet, P.; Ricci, V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 2012, 20, 165–174. [Google Scholar] [CrossRef]
- Leyton, D.L.; Rossiter, A.E.; Henderson, I.R. From self sufficiency to dependence: Mechanisms and factors important for autotransporter biogenesis. Nat. Rev. Microbiol. 2012, 10, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Woo, H.; Park, M.; Rhee, K.-J.; Moon, C.; Lee, D.; Seo, W.D.; Kim, J.B. Cyanidin 3-O-glucoside reduces Helicobacter pylori VacA-induced cell death of gastric KATO III cells through inhibition of the SecA pathway. Int. J. Med. Sci. 2014, 11, 742–747. [Google Scholar] [CrossRef]
- Lee, M.H.; Yang, J.Y.; Cho, Y.; Woo, H.J.; Kwon, H.J.; Kim, D.H.; Park, M.; Moon, C.; Yeon, M.J.; Kim, H.W.; et al. Inhibitory effects of menadione on Helicobacter pylori growth and Helicobacter pylori-induced inflammation via NF-κB inhibition. Int. J. Mol. Sci. 2019, 20, 1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Lee, P.; Kim, J.B. Effect of Evodiae fructus methanol extract on virulence-related genes’ expression of Helicobacter pylori. Korean J. Clin. Lab. Sci. 2019, 51, 316–322. [Google Scholar] [CrossRef]
- Nguyen, N.V.T.; Lee, K.R.; Lee, Y.J.; Choi, S.; Kang, J.S.; Mar, W.; Kim, K.H. Chiral high-performance liquid chromatographic separation of evodiamine enantiomers and rutaecarpine, isolated from Evodiae fructus. J. Pharm. Biomed. Anal. 2013, 81–82, 151–159. [Google Scholar] [CrossRef]
- Chiou, W.-F.; Chou, C.-J.; Shum, A.Y.-C.; Chen, C.-F. The vasorelaxant effect of evodiamine in rat isolated mesenteric arteries: Mode of action. Eur. J. Pharmacol. 1992, 215, 277–283. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nakano, Y.; Kizaki, M.; Hoshikuma, K.; Yokoo, Y.; Kamiya, T. Capsaicin-like anti-obese activities of evodiamine from fruits of Evodia rutaecarpa, a vanilloid receptor agonist. Planta Med. 2001, 67, 628–633. [Google Scholar] [CrossRef]
- Shin, Y.-W.; Bae, E.-A.; Cai, X.F.; Lee, J.J.; Kim, D.-H. In vitro and in vivo antiallergic effect of the fructus of Evodia rutaecarpa and its constituents. Biol. Pharm. Bull. 2007, 30, 197–199. [Google Scholar] [CrossRef]
- Chiou, W.-F.; Sung, Y.-J.; Liao, J.-F.; Shum, A.Y.-C.; Chen, C.-F. Inhibitory effect of dehydroevodiamine and evodiamine on nitric oxide production in cultured murine macrophages. J. Nat. Prod. 1997, 60, 708–711. [Google Scholar] [CrossRef]
- Ogasawara, M.; Matsubara, T.; Suzuki, H. Inhibitory effects of evodiamine on in vitro invasion and experimental lung metastasis of murine colon cancer cells. Biol. Pharm. Bull. 2001, 24, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-H.; Pan, S.-L.; Guh, J.-H.; Chang, Y.-L.; Pai, H.-C.; Lin, C.-H.; Teng, C.-M. Antitumor mechanism of evodiamine, a constituent from Chinese herb Evodiae fructus, in human multiple-drug resistant breast cancer NCI/ADR-RES cells in vitro and in vivo. Carcinogenesis 2005, 26, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.-F.; Yu, C.-H.; Pu, H.-F.; Hsu, J.-M.; Chen, M.-J.; Wang, P.S. Anti-proliferative effects of evodiamine on human prostate cancer cell lines DU145 and PC3. J. Cell. Biochem. 2007, 101, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kersulyte, D.; Mukhopadhyay, A.K.; Velapatiño, B.; Su, W.; Pan, Z.; Garcia, C.; Hernandez, V.; Valdez, Y.; Mistry, R.S.; Gilman, R.H.; et al. Differences in genotypes of Helicobacter pylori from different human populations. J. Bacteriol. 2000, 182, 3210–3218. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Cornaggia, M.; Valentin, M. Helicobacter pylori, pre-neoplastic changes, gastric cancer: A point of view. Eur. J. Gastroenterol. Hepatol. 1999, 11, 357–359. [Google Scholar] [CrossRef]
- Peleteiro, B.; Bastos, A.; Ferro, A.; Lunet, N. Prevalence of Helicobacter pylori infection worldwide: A systematic review of studies with national coverage. Dig. Dis. Sci. 2014, 59, 1698–1709. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Zhou, L.-Y.; Song, Z.-Q.; Zhang, J.-Z.; He, L.-H.; Ding, Y. Primary antibiotic resistance of Helicobacter pylori strains isolated from patients with dyspeptic symptoms in Beijing: A prospective serial study. World J. Gastroenterol. 2015, 21, 2786–2792. [Google Scholar] [CrossRef]
- Wu, I.-T.; Chuah, S.-K.; Lee, C.-H.; Liang, C.-M.; Lu, L.-S.; Kuo, Y.-H.; Yen, Y.-H.; Hu, M.-L.; Chou, Y.-P.; Yang, S.-C.; et al. Five-year sequential changes in secondary antibiotic resistance of Helicobacter pylori in Taiwan. World J. Gastroenterol. 2015, 21, 10669–10674. [Google Scholar] [CrossRef]
- Ghotaslou, R.; Leylabadlo, H.E.; Asl, Y.M. Prevalence of antibiotic resistance in Helicobacter pylori: A recent literature review. World J. Methodol. 2015, 5, 164–174. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Chang, M.-C.; Chen, C.-S.; Lin, H.-C.; Tsai, H.-P.; Yang, C.-C.; Yang, C.-H.; Lin, C.-M. Topoisomerase I inhibitor evodiamine acts as an antibacterial agent against drug-resistant Klebsiella pneumoniae. Planta Med. 2013, 79, 27–29. [Google Scholar] [CrossRef]
- Choi, Y.H.; Shin, E.M.; Kim, Y.S.; Cai, X.F.; Lee, J.J.; Kim, H.P. Anti-inflammatory principles from the fruits of Evodia rutaecarpa and their cellular action mechanisms. Arch. Pharmacal Res. 2006, 29, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hua, Q.; Li, N.; Zhao, M.; Cui, Y. Protective effects of evodiamine against LPS-induced acute kidney injury through regulation of ROS-NF-κB-mediated inflammation. Evid. Based Complement. Altern. Med. 2019, 2019, 2190847. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, N.; Ishii, E.; Tominaga, K.; Tezuka, Y.; Nagaoka, T.; Kadota, S.; Kuroki, T.; Yano, I. Highly selective antibacterial activity of novel alkyl quinolone alkaloids from a Chinese herbal medicine, Gosyuyu (Wu-Chu-Yu), against Helicobacter pylori in vitro. Microbiol. Immunol. 2000, 44, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kling, A.; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C.; et al. Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Zhu, J.-H.; Wang, B.-W.; Pan, M.; Zeng, Y.-N.; Rego, H.; Javid, B. Rifampicin can induce antibiotic tolerance in mycobacteria via paradoxical changes in rpoB transcription. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Rho, T.C.; Bae, E.-A.; Kim, D.-H.; Oh, W.K.; Kim, B.Y.; Ahn, J.S.; Lee, H.S. Anti-Helicobacter pylori activity of quinolone alkaloids from Evodiae Fructus. Biol. Pharm. Bull. 1999, 22, 1141–1143. [Google Scholar] [CrossRef]
- Fan, X.; Gunasena, H.; Cheng, Z.; Espejo, R.; Crowe, S.E.; Ernst, P.B.; Reyes, V.E. Helicobacter pylori Urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J. Immunol. 2000, 165, 1918–1924. [Google Scholar] [CrossRef]
- Beswick, E.J.; Pinchuk, I.V.; Minch, K.; Suarez, G.; Sierra, J.C.; Yamaoka, Y.; Reyes, V.E. The Helicobacter pylori urease B subunit binds to CD74 on gastric epithelial cells and induces NF-κB activation and interleukin-8 production. Infect. Immun. 2006, 74, 1148–1155. [Google Scholar] [CrossRef]
- Frick-Cheng, A.E.; Pyburn, T.M.; Voss, B.J.; McDonald, W.H.; Ohi, M.D.; Cover, T.L. Molecular and structural analysis of the Helicobacter pylori cag type IV secretion system core complex. mBio 2016, 7, e02001-15. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.; Flores-Encarnación, M.; Aguilar-Gutiérrez, G.R. Functional interaction and structural characteristics of unique components of Helicobacter pylori T4SS. FEBS J. 2017, 284, 3540–3549. [Google Scholar] [CrossRef]
- Redzej, A.; Ukleja, M.; Connery, S.; Trokter, M.; Felisberto-Rodrigues, C.; Cryar, A.; Thalassinos, K.; Hayward, R.D.; Orlova, E.V.; Waksman, G. Structure of a VirD4 coupling protein bound to a VirB type IV secretion machinery. EMBO J. 2017, 36, 3080–3095. [Google Scholar] [CrossRef] [PubMed]
- Terradot, L.; Waksman, G. Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J. 2011, 278, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L.T.; Mendz, G.L.; Hazell, S.L. Overview. In Helicobacter pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Schmausser, B.; Josenhans, C.; Endrich, S.; Suerbaum, S.; Sitaru, C.; Andrulis, M.; Brändlein, S.; Rieckmann, P.; Müller-Hermelink, H.K.; Eck, M. Downregulation of CXCR1 and CXCR2 expression on human neutrophils by Helicobacter pylori: A new pathomechanism in H. pylori infection? Infect. Immun. 2004, 72, 6773–6779. [Google Scholar] [CrossRef]
- Kim, J.-M.; Kim, K.-M.; Park, E.-H.; Seo, J.-H.; Song, J.-Y.; Shin, S.-C.; Kang, H.-L.; Lee, W.-K.; Cho, M.-J.; Rhee, K.-H.; et al. Anthocyanins from black soybean inhibit Helicobacter pylori-induced inflammation in human gastric epithelial AGS cells. Microbiol. Immunol. 2013, 57, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Suzuki, M.; Mori, M.; Kitahora, T.; Yokoyama, H.; Miura, S.; Hibi, T.; Ishii, H. Augmented levels of gastric mucosal leucocyte activation by infection with cagA gene-positive Helicobacter pylori. J. Gastroenterol. Hepatol. 1998, 13, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lim, J.W.; Kim, K.H. Helicobacter pylori-induced expression of interleukin-8 and cyclooxygenase-2 in AGS gastric epithelial cells: Mediation by nuclear factor-κB. Scand. J. Gastroenterol. 2001, 36, 706–716. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, J.S.; Lee, J.Y.; Kim, Y.-J.; Youn, H.-J.; Kim, I.Y.; Chee, Y.J.; Oh, Y.-K.; Kim, N.; Jung, H.C.; et al. Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-κB activation. Infect. Immun. 2007, 75, 3373–3381. [Google Scholar] [CrossRef]
- Hisatsune, J.; Nakayama, M.; Isomoto, H.; Kurazono, H.; Mukaida, N.; Mukhopadhyay, A.K.; Azuma, T.; Yamaoka, Y.; Sap, J.; Yamasaki, E.; et al. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-κB activation. J. Immunol. 2008, 180, 5017–5027. [Google Scholar] [CrossRef]
- Meyer-Ter-Vehn, T.; Covacci, A.; Kist, M.; Pahl, H.L. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J. Biol. Chem. 2000, 275, 16064–16072. [Google Scholar] [CrossRef]
- Lee, M.H.; Kwon, H.J.; Kim, D.H.; Yang, J.Y.; Cho, Y.; Woo, H.T.; Yeon, M.J.; Park, M.; Moon, C.; Kim, S.H.; et al. Kinetin inhibits growth of Helicobacter pylori by down-regulation of replication genes. Int. J. Clin. Exp. Med. 2017, 10, 795–801. [Google Scholar]
- Yeon, M.J.; Lee, M.H.; Kim, D.H.; Yang, J.Y.; Woo, H.J.; Kwon, H.J.; Moon, C.; Kim, S.-H.; Kim, J.-B. Anti-inflammatory effects of Kaempferol on Helicobacter pylori-induced inflammation. Biosci. Biotechnol. Biochem. 2019, 83, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Boonjakuakul, J.K.; Canfield, D.R.; Solnick, J.V. Comparison of Helicobacter pylori virulence gene expression in vitro and in the Rhesus Macaque. Infect. Immun. 2005, 73, 4895–4904. [Google Scholar] [CrossRef] [PubMed]
- Tharmalingam, N.; Kim, S.-H.; Park, M.; Woo, H.J.; Kim, H.W.; Yang, J.Y.; Rhee, K.-J.; Kim, J.B. Inhibitory effect of piperine on Helicobacter pylori growth and adhesion to gastric adenocarcinoma cells. Infect. Agents Cancer 2014, 9, 43. [Google Scholar] [CrossRef]
- Clayton, C.; Kleanthous, K.; Tabaqchali, S. Detection and identification of Helicobacter pylori by the polymerase chain reaction. J. Clin. Pathol. 1991, 44, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef]
- Sharma, S.A.A.; Tummuru, M.K.; Blaser, M.J.; Kerr, L.D. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J. Immunol. 1998, 160, 2401–2407. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evodiamine Concentration (μM) | Number of Strains (%) | |
|---|---|---|
| ≤5 | 7 | (14%) |
| 10 | 10 | (20%) |
| 20 | 21 | (42%) |
| 40 | 7 | (14%) |
| ≥80 | 5 | (10%) |
| Total | 50 | (100%) |
| Primers | Sequences (5′-3′) | Product Length (bp) | Annealing Temperature (°C) | Cycles | Reference | |
|---|---|---|---|---|---|---|
| Forward | Reverse | |||||
| DnaA | GGGCATGACTTAGCGGTTA | TTAACGAATTGCACGCCAAC | 128 | 55 | 27 | [62] |
| DnaB | AATGGGCCGTTTATCGTCTC | CAAATCCGCTTGCAACTACG | 231 | 55 | 27 | |
| DnaE | AATCCACCGGCTCCAAATAC | GCCAAACAAGTGTGGGAGTA | 184 | 55 | 27 | |
| DnaN | GTTAGCGGTGGTTGAAAACG | CGGTTTCGCTATGCTCAGAA | 233 | 55 | 27 | |
| DnaQ | CGCATGAAGCTTTGCAAGAA | GCATAGGCTCTATGGCTGAC | 244 | 55 | 27 | |
| GyrA | GTGCATAGGCGTATTTT | CATTCTGGCTTCAGTGTAACG | 246 | 52 | 25 | |
| RpoA | AGCGACACGTCTTCAGTAAC | ACAGCACCTTTGATCCCATC | 224 | 55 | 22 | [65] |
| RpoB | TTTAGGTAAGCGCGTGGATT | AATCAGCTTTGGATGGAACG | 301 | 59 | 24 | |
| RpoD | TCATCATCATTGCCGACTGG | GTCATGCGCAAACACATTCA | 152 | 55 | 26 | |
| RpoN | GCCCTTGAAATCGTGCTTAC | ATGATGAGAGCTACCCGACA | 250 | 55 | 27 | |
| UreA | GCCAATGGTAAATTAGTT | CTCCTTAATTGTTTTTAC | 411 | 40 | 20 | [66] |
| UreB | TCTATCCCTACCCCACAACC | CCATCCACGAACACATGGTA | 252 | 50 | 21 | |
| CagA | GTCATAATGGCATAGAACCTGAA | ATTCCCTAGGGCGTCTAAATAA | 407 | 59 | 21 | [63] |
| VirB2 | CAGTCGCCTGACCTCTTTTGA | CGGTCACCAGTCCTGCAAC | 156 | 62 | 25 | |
| VirB4 | GTTATAGGGGCAACCGGAAG | TTGAACGCGTCATTCAAAGC | 449 | 62 | 37 | |
| VirB5 | TACAAGCGTCTGTGAAGCAG | GACCAACCAACAAGTGCTCA | 436 | 62 | 30 | |
| VirB6 | CCTCAACACCGCCTTTGGTA | TAGCCGCTAGCAATCTGGTG | 225 | 62 | 25 | |
| VirB7 | GATTACGCTCATAGGCGATGC | TGGCTGACTTCCTTGCAACA | 202 | 62 | 25 | |
| VirB8 | GTTGATCCTTGCGATCCCTCA | CGCCGCTGTAACGAGTATTG | 218 | 62 | 25 | |
| VirB9 | GCATGTCCTCTAGTCGTTCCA | TATCGTAGATGCGCCTGACC | 269 | 62 | 25 | |
| VirD4 | CCGCAAGTTTCCATAGTGTC | GCGAGTTGGGAAACTGAAGA | 263 | 62 | 25 | |
| SecA | AAAAATTTGACGCTGTGATCC | CCCCCAAGCTCCTTAATTTC | 274 | 47 | 27 | |
| VacA | AAACGACAAGAAAGAGATCAGT | CCAGCAAAAGGCCCATCAA | 291 | 57 | 22 | [64] |
| Efp | GGCAATTTGGATGAGCGAGCTC | CTTCACCTTTTCAAGATACTC | 559 | 59 | 23 | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.Y.; Kim, J.-B.; Lee, P.; Kim, S.-H. Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation. Int. J. Mol. Sci. 2021, 22, 3385. https://doi.org/10.3390/ijms22073385
Yang JY, Kim J-B, Lee P, Kim S-H. Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation. International Journal of Molecular Sciences. 2021; 22(7):3385. https://doi.org/10.3390/ijms22073385
Chicago/Turabian StyleYang, Ji Yeong, Jong-Bae Kim, Pyeongjae Lee, and Sa-Hyun Kim. 2021. "Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation" International Journal of Molecular Sciences 22, no. 7: 3385. https://doi.org/10.3390/ijms22073385
APA StyleYang, J. Y., Kim, J.-B., Lee, P., & Kim, S.-H. (2021). Evodiamine Inhibits Helicobacter pylori Growth and Helicobacter pylori-Induced Inflammation. International Journal of Molecular Sciences, 22(7), 3385. https://doi.org/10.3390/ijms22073385