1. Introduction
Adenoviruses are double-stranded DNA viruses that deliver their large 35 kbp genome to the nuclei of nondividing cells. The intracellular trafficking of adenoviruses has been studied extensively, but the mechanism that facilitates the final step of import of adenovirus DNA through the nuclear pore is not well-understood [
1]. Unravelling the nuclear transport mechanism at the molecular level will provide insight into the import requirements of large DNA molecules and could provide a valuable blueprint for the design of synthetic pharmaceutical gene delivery systems. Adenoviral DNA is assumed to enter the nucleus as a complex with some or all of the viral core proteins [
2,
3]. However, which of these components is essential for import is not clear. The core of the adenovirus has three major proteins—pV, pVII and adenovirus terminal protein (TP)—that collectively compact and protect its genome. One molecule of TP is covalently attached to each DNA strand at the 5′ end linking to the phosphate group in the DNA backbone. It is first expressed within a host cell as preterminal protein (pTP), and subsequently translocated to the nucleus where it is coupled to newly replicated viral DNA. pTP is later cleaved by adenovirus proteases to produce TP as each viral particle matures. This unique covalent DNA–protein link is established at pTP Ser–580 (referred to in this study as Ser–562, see experimental procedures below). pTP is critical for adenovirus polymerase priming and subsequent adenovirus replication [
4,
5]. In the infecting virus, the viral DNA appears to use its attached TPs as nuclear matrix anchors [
6], thus enhancing its extrachromosomal stability and improving adenovirus gene expression. Previous work indicated that major core protein pVII, a DNA–condensing protein, appears to play an essential role in DNA delivery [
7]. Whether TP assists the incoming viral DNA during nuclear entry is not yet clear.
Studies of adenovirus TP have been limited, and the relationship between adenovirus TP and its nuclear entry is also not well-defined. An early study identified a positively charged motif of pTP that contains a nuclear localisation signal (NLS), PV(R)
6VP. This motif was reported to be essential for adenovirus pTP translocation into the nucleus by observing codelivery of the associated viral polymerase [
8]. The positively charged motif identified is upstream of a proline-rich area within the pTP sequence. The removal of the region containing the PV(R)
6VP sequence inhibited TP nuclear transport. The study’s authors concluded that the TP NLS resembled the prototypical simian vacuolating virus, (SV40) large T–antigen sequence and was necessary for pTP delivery to the nucleus.
NLSs are drivers of protein transport across the nucleus [
9]. Their presence has also been implicated in the delivery of large DNA molecules [
10]. NLSs resemble the SV40 T–antigen sequence (PKKKRKV) and typically bind with importin α (Imp α) protein adaptors. Complexed Imp α then binds to importin β (Imp β) through its conserved importin β binding domain (IBB) domain [
11] to deliver the protein cargo. Other forms of NLSs may translocate to the nucleus without binding to Imp α. This nuclear delivery can be achieved by directly binding to Imp β [
12] or one of its >20 homologues [
13]. NLSs can either be monopartite, carrying one essential positively charged region or bipartite, such as the classical nucleoplasmin signal [
14], with two separate but interdependent positively charged peptide motifs separated by a 10–12 bp spacer. For optimal nuclear localisation, both regions are required.
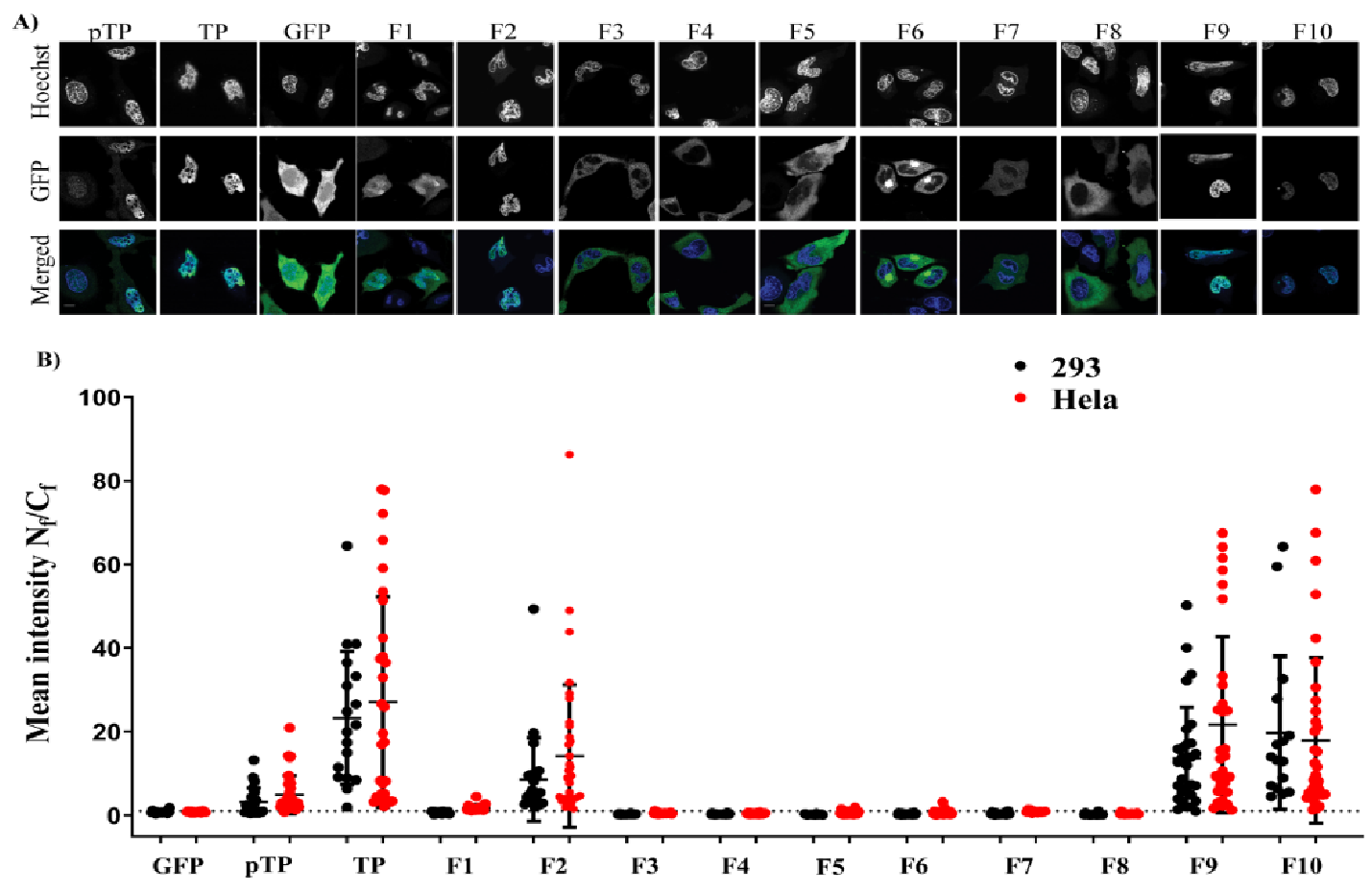
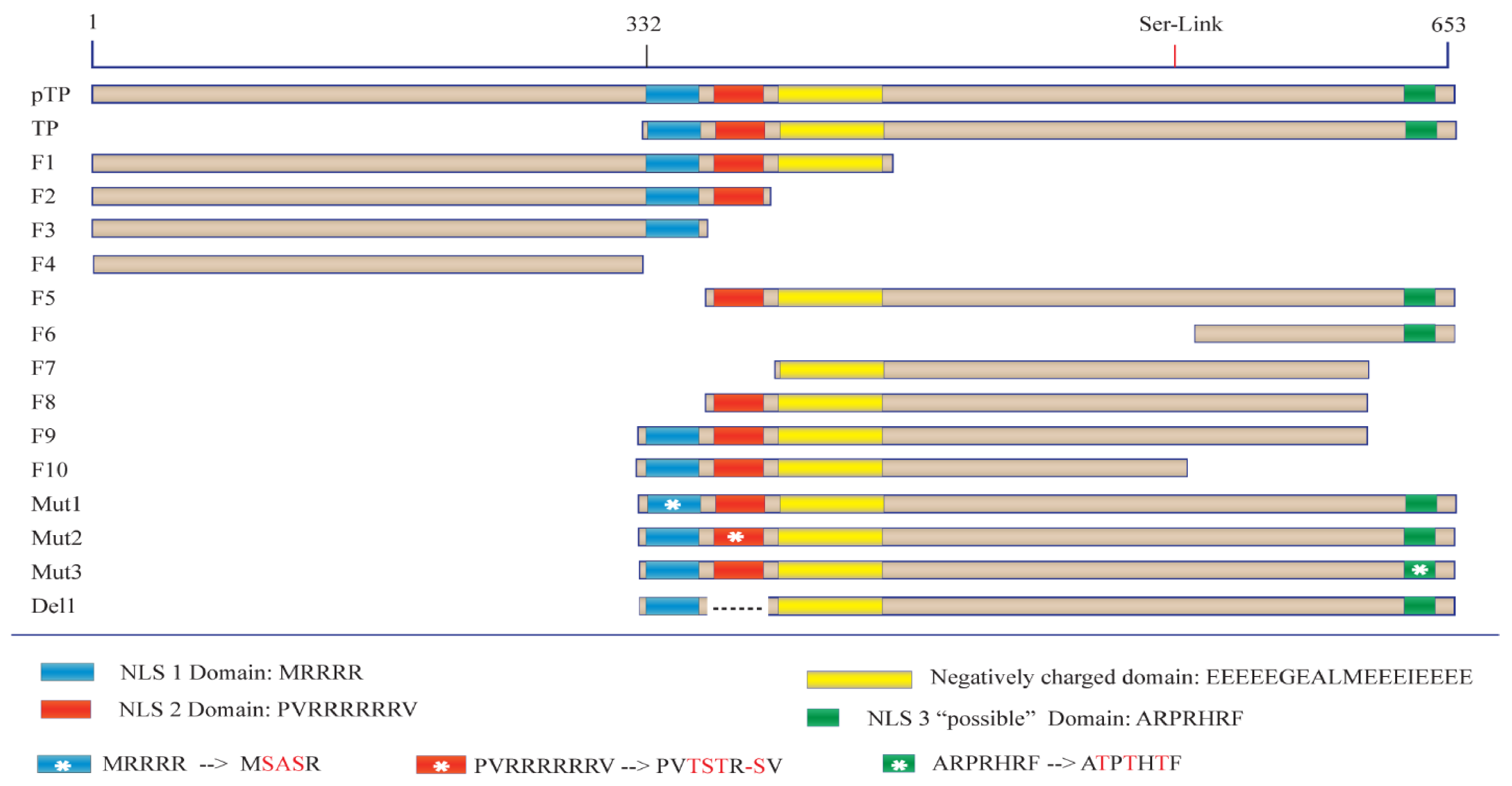
Before the role of TP in viral DNA entry can be tested, a detailed understanding of the protein sequences required for nuclear localisation must first be elucidated. We began this study by examining various regions of TP utilising a series of engineered GFP fusion proteins. This strategy allowed us to compare the nuclear localisation of each fusion and led us to identify a key bipartite signal essential for fully efficient nuclear entry of TP. Furthermore, the current study examines the localisation of bacterially expressed TP using biochemical and in vitro assays of nuclear delivery. Finally, in this study, we explore the form of nuclear interaction that led to TP nuclear localisation and identify Imps that could be involved in the nuclear entry of TP.
3. Discussion
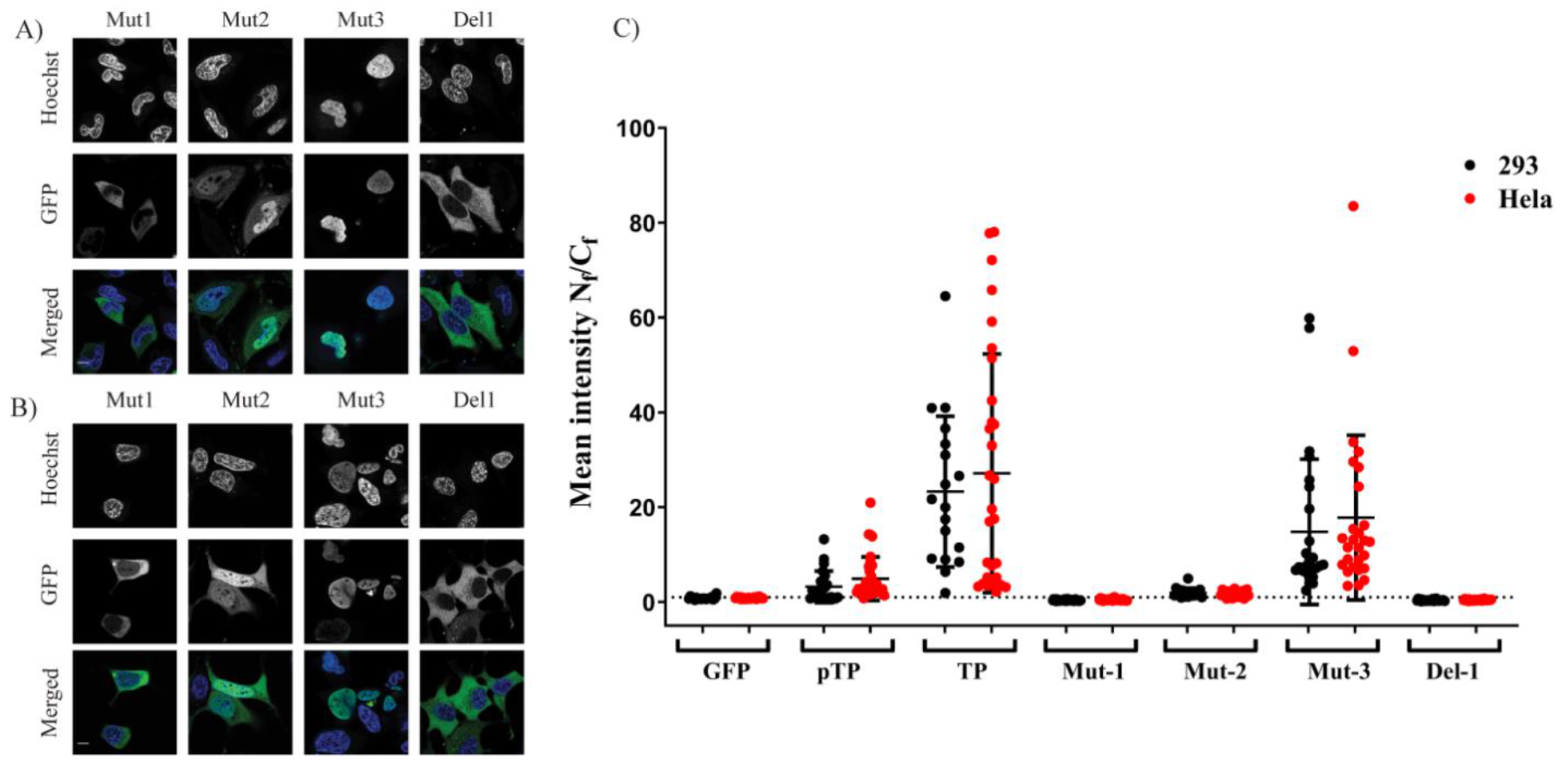
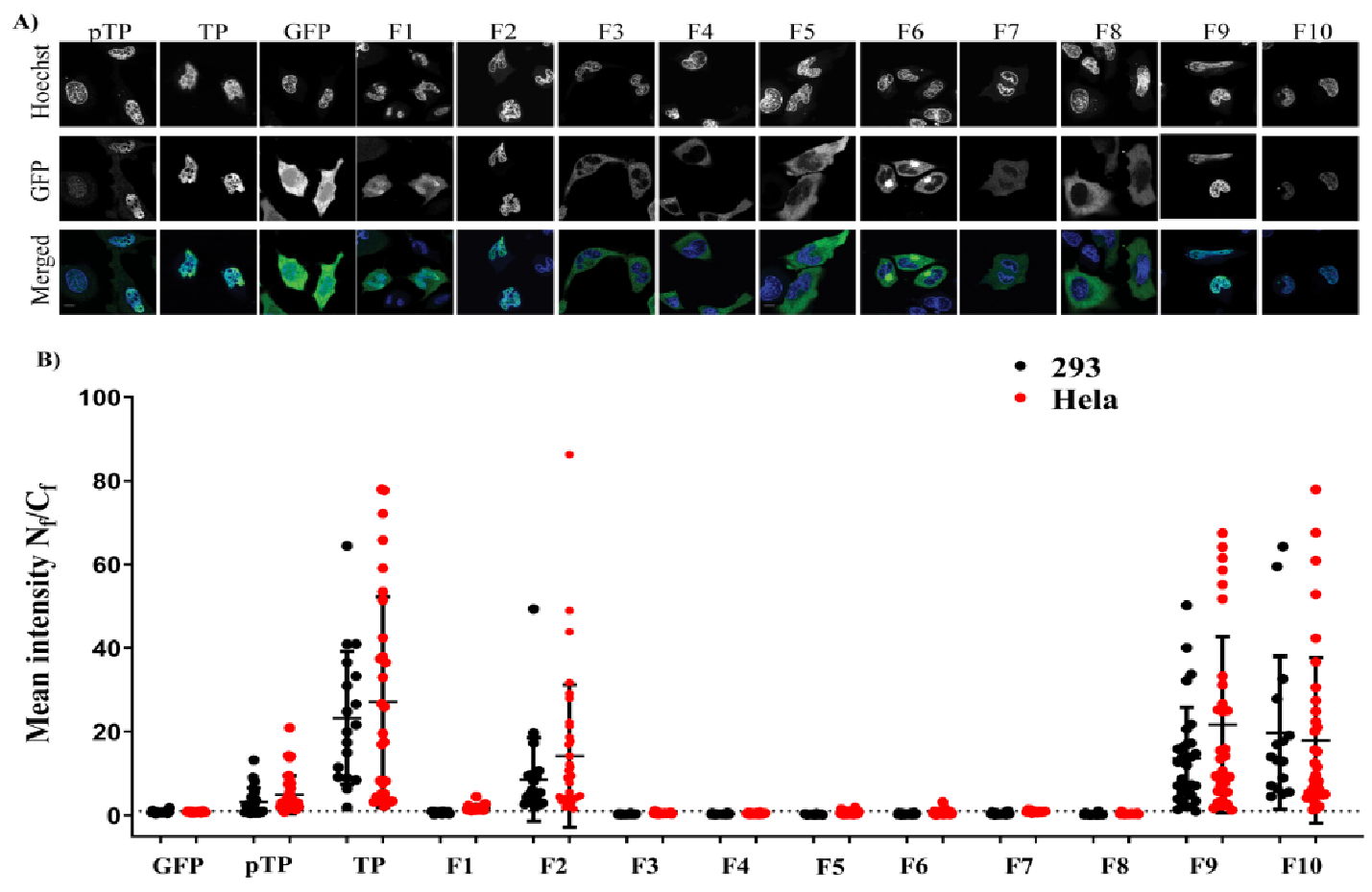
In this study, we applied bioengineering and biochemical methods to study adenovirus terminal protein nuclear localisation and the mechanism of this localisation. Although adenoviruses are a well-characterised DNA virus model, little attention was placed on studying the intracellular trafficking of adenoviral TP. Sequence analysis of TP led us to propose three potential NLSs (described in this manuscript as NLS1, NLS2 and NLS3).
In the first part of this study, we found that TP and its precursor (pTP) localise in the nucleus. This localisation was independent of the viral polymerase [
8], despite the cell-type used. In the first bioengineered fragment, which exhibited inhibition of nuclear localisation, a motif rich in negatively charged residues (EEEEGEALMEEEIEEEEE) remained downstream of NLS
2. Although the negatively charged domain is present in full-length TP, it did not impact on nuclear translocation as long as it was not the last motif in the C–terminal flank of the protein (see, e.g., F9 and F10). It is possible that the negatively charged domain interfered with the function of NLS
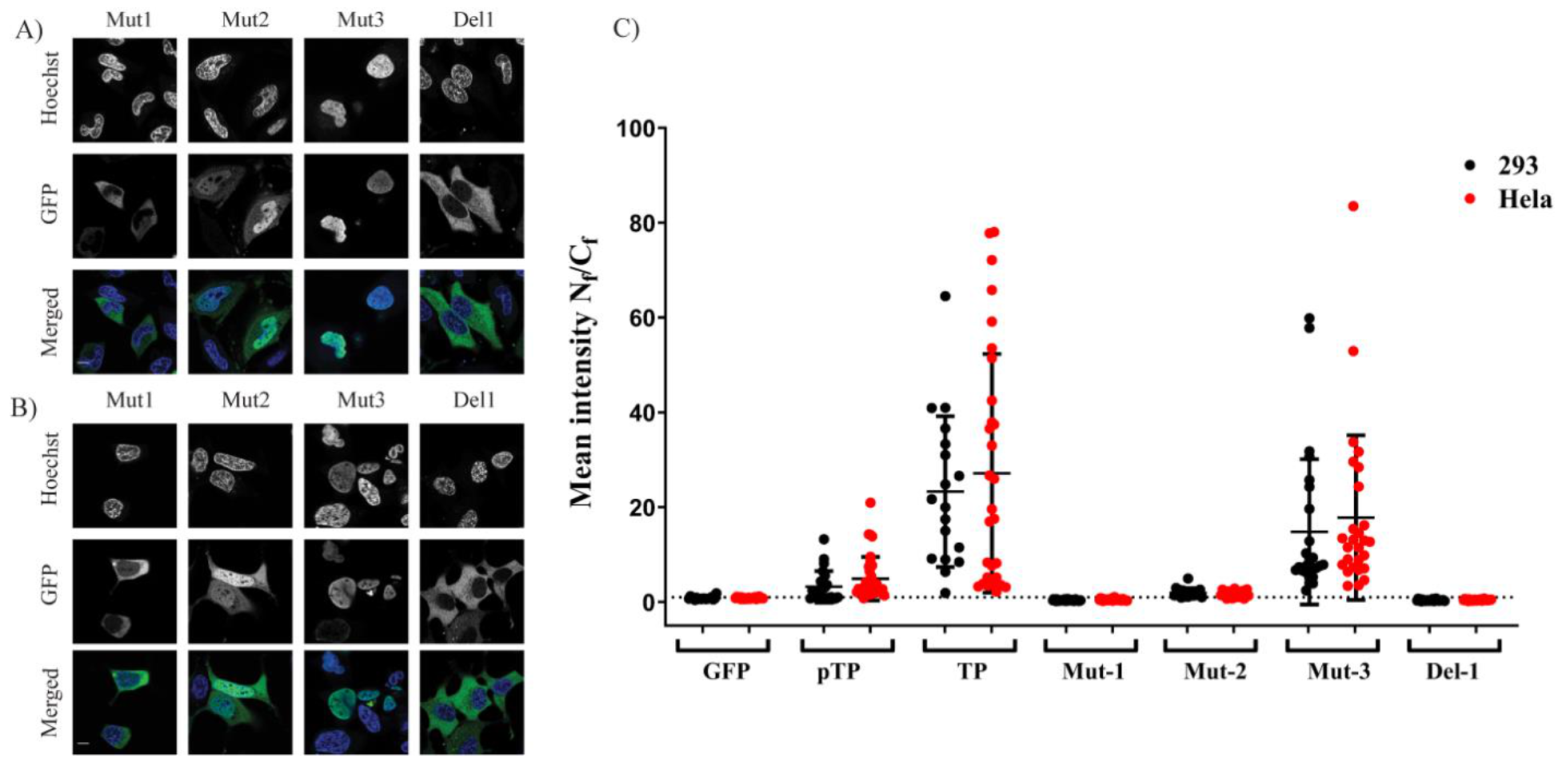
2 when it lacked a structural constraint provided by the full downstream structure at the C–terminus of the motif. The second part of the study identified the NLS required for nuclear entry of TP. Further fragmentation and mutation studies indicated the requirement of a bipartite NLSs for nuclear localisation of transfected TP fragments. The use of mutations and NLS selective deletion validated that a bipartite sequence, observed in the fragmentation study (F1–F10), was necessary for full NLS activity, not just the result of the structural constraints that result from the removal of motifs from the TP sequence. The data obtained from comprehensively mutating and fragmenting TP indicate that the protein makes use of a more complicated mechanism than the monopartite nuclear translocation mechanism described previously [
8].
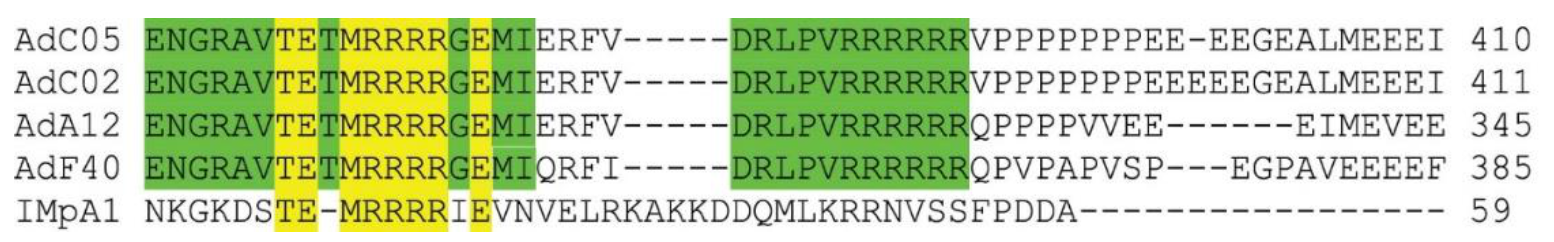
The difference between our results and the previous study have been revealed by the specific fragmentation approach used in this study. In the previous study, the fragment that was removed by the researchers was a much longer sequence from the TP open reading frame (ORF), resulting in deletion of the accompanying NLS
1. Interestingly, the NLS
1 sequence (
Figure 4) shows a striking similarity to the IBB domain (an amino acid domain part of Imp α adaptor) in that it facilitates the binding of Imp α to Imp β [
11]. Bipartite NLSs have been reported to bind to the Imp α/β heterodimer [
21,
22]. Alternatively, a nonclassical NLS has been identified which binds directly to Imp β, such as the NLS found in parathyroid hormone-related protein (PTHrP). However, the latter protein is not considered to include a prototypical bipartite NLS with two positively charged amino acid patches [
12]. TP has an NLS that is similar to the Imp α IBB domain (
Figure 4), and we show in this study that this signal is essential for nuclear entry (
Figure 3A–C). The IBB domain found in Imp α binds to Imp β to form a high-affinity α/β heterodimer [
23] before shuttling to the nucleus. The idea that the NLS of TP represents a class of bipartite signals capable of directly interacting with Imp β is interesting since this form of binding is not typical of bipartite NLSs. Whether the bipartite signal within TP can bind to Imp β warranted us performing further investigations (see below).
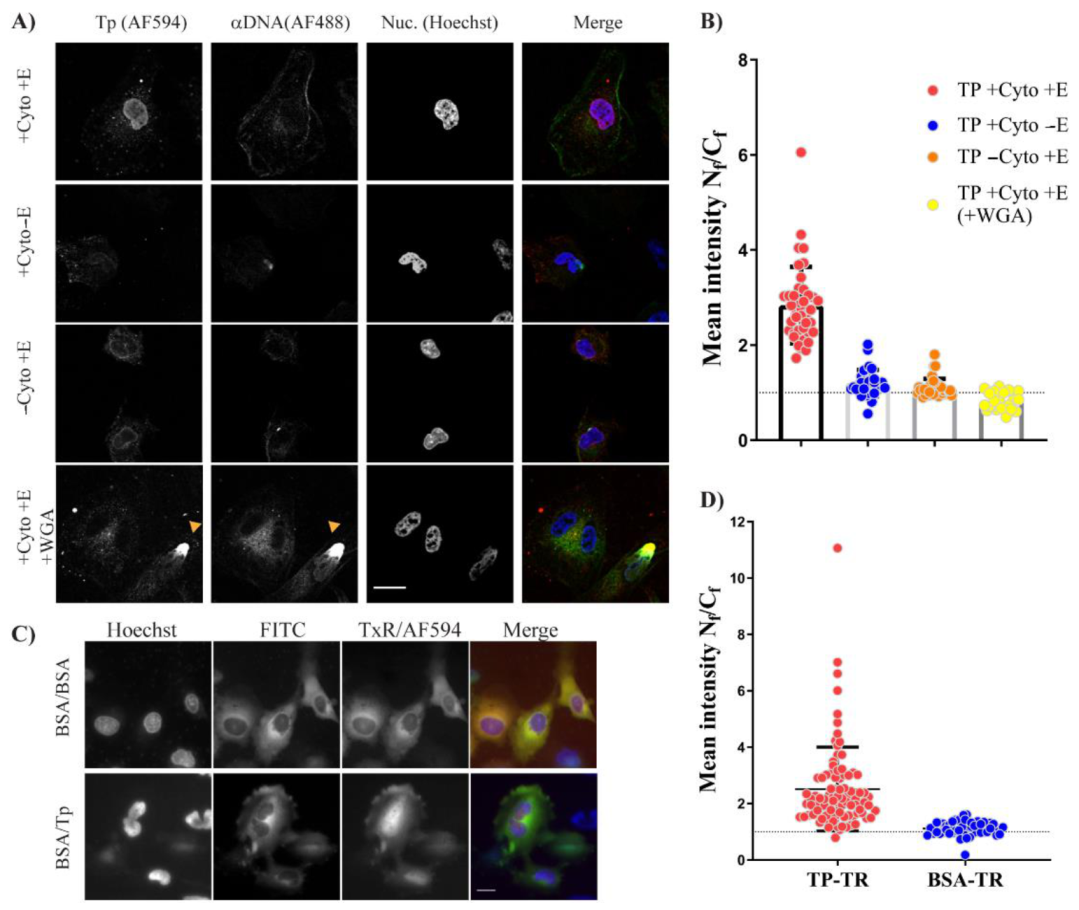
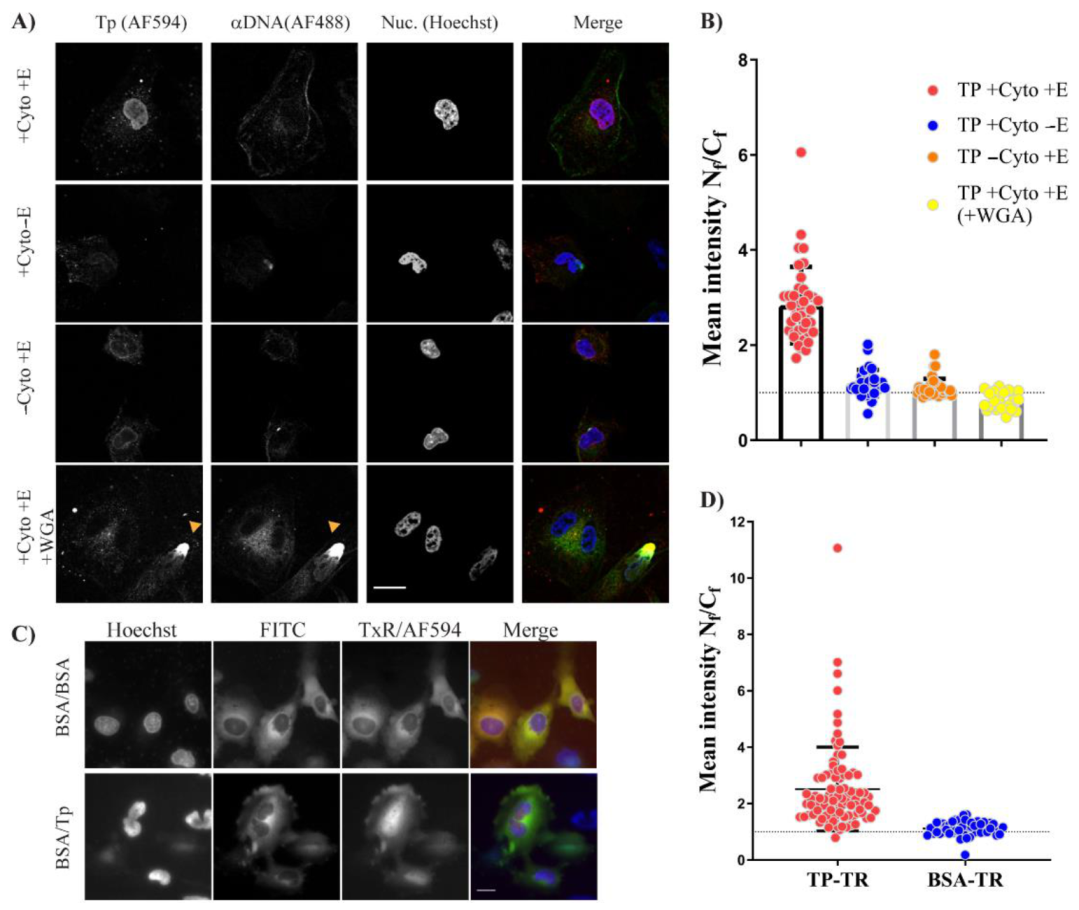
After successful autologous expression, we found that bacterially expressed TP–Trx required active transport to reach the nucleus. We used the digitonin permeabilisation assay (DPA), a widely used assay to investigate the nuclear localisation of proteins. In this assay, we incorporated an anti–DNA antibody which was essential to avoid false-positives due to possible nuclear membrane damage by digitonin treatment. The requirement of energy and cytosolic factors suggests that the TP–Trx can enter the nucleus by interaction with shuttling proteins and the shuttling requires energy to be completed. The nuclear localisation of TP–Trx–AF594 after cytoplasmic microinjection in living cells supports the active transport hypothesis. While this access is robust and statistically significant (
Figure 6D), some signal in the cytoplasm remained. An NES signal on TP which may have allowed some continued export of the protein could explain this observation. However, our results following leptomycin B treatment, an inhibitor of exportin 1, showed no impact on nuclear entry of TP compared to untreated control (
Figure 7B). The combined results of DPA and microinjection clearly indicate that access to the nucleus from the cytoplasm is an active process requiring energy.
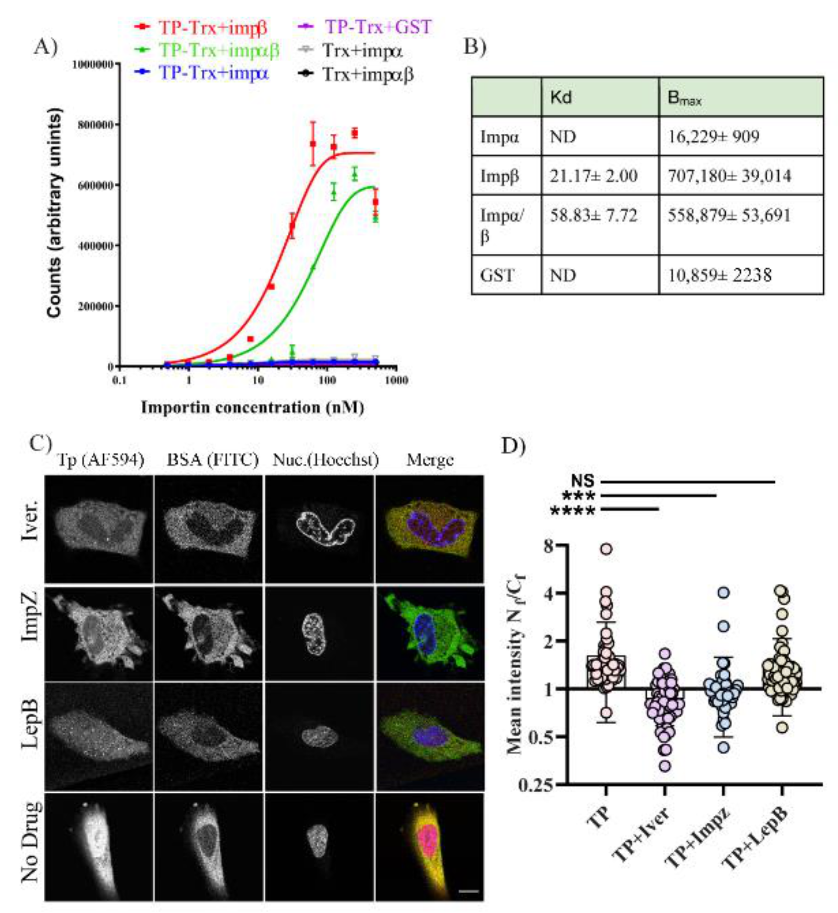
Since active transport requires interaction with a nuclear shuttling factor, we considered which of the cellular factors could be utilised by TP. Our observations that TP can strongly bind Imp β1 and Imp α/β1 were consistent in both the in vitro AlphaScreen assay (
Figure 7A) and microinjection in living cells. In living cells, inhibitors of both Imp α/β1 and Imp β1 significantly lowered nuclear accumulation of TP (
Figure 7B,D). Although our results strongly indicate that TP can bind and use Imp α/β1 and Imp β1 for nuclear transport, we cannot exclude other cytosolic factors that could assist TP in its nuclear accumulation. Using inhibitors such as ivermectin and importazole, we show significant but incomplete inhibition. We could not use a higher concentration of these drugs as we noticed significant toxicity when attempting microinjection in the presence of higher concentrations of these compounds. As expected, we did not observe strong binding of TP to Imp α in the AlphaScreen assay. This can be explained by the well-known autoinhibition of the Imp α binding site for NLSs in the absence of Imp β [
24].
Our study offers an opportunity to study antiviral interventions. Indeed, TP entry to the nucleus is crucial for viral replication and infection [
5]. Our study also highlights the possibility of TP involvement in incoming viral DNA entry. Intervening with TP entry to the nucleus can be useful to mitigate adenoviral infections. Adenoviruses are common viruses with respiratory, neurological, eye and gastrointestinal infections [
25]. In our study, the use of ivermectin and importazol helped reduce TP entry to the nucleus. Both these drugs and others can be tested on adenoviruses, as can the impact of drugs on viral entry and replication. Our work shows intimate and high-affinity binding between Importin αβ or Importin β. However, structural studies can be used to probe whether TP–importins interaction is unique or whether it can be disrupted by other drugs that are known to affect importin trafficking or interaction.
The results from this work provide an essential foundation to answer whether TP is needed for nuclear entry of adenovirus in the future. The current study focuses on identifying and studying the nuclear localisation sequences of TP and its biochemical interactions with host importins. This aim provides a basic and comprehensive understanding of the mechanism of entry of this protein, prior to studying the TP in the context of viral entry. Future work involving the study of viral particles with mutant TP will be important to clarify the rule of TP in viral entry. However, studying adenoviral TP in the context of nuclear pore entry cycle may pose certain technical challenges. Nuclear localisation of newly expressed TP is a vital first step of adenovirus replication and generating viral particles. Mutations to prevent entry can stop the generation of reporter viral particles for studying the phenomenon of viral nuclear entry. Therefore, development of techniques that circumvent such challenge will be needed.
Interestingly, in some bacteriophage and
Streptomyces spp. the relevant terminal proteins possess NLSs [
26,
27] that can enhance gene expression when transfected in mammalian cells. It is yet unclear how the terminal proteins drive nuclear translocation in eukaryotic cells and whether they interact with mammalian nuclear Imps during viral nuclear delivery. This will be an interesting topic of investigation in the future. Our work proposes a new question: is TP involved in adenoviral trafficking to the nucleus? Intracellular viral trafficking is thought to be carried out by a hexon, which the virus uses to dock into the nuclear pore [
2]. However, this docking may be incomplete [
1]. Previous work showed some involvement of the condensing protein, pVII, in viral delivery across the nucleus [
7]. However, in that study, TP was also present in the incoming viral DNA. Tp is present only as two protein copies per viral particle, which makes its identification and localisation difficult. Recently, a biotechnological approach to use TP–DNA as a cloning vector helped produce higher titre of adenovirus after transfection into permissive cell lines [
28].
In this study, we used cellular and molecular biology approaches to study TP protein localisation to the nucleus, an essential step in the initiation of adenovirus replication inside the nucleus. Our results show that TP contains a bipartite NLS. Both NLSs must be present for nuclear entry. This entry requires shuttling factors and energy. Finally, the use of Imp β and Imp α/ β heterodimer confirms that potentially either route of nuclear entry is possible. Our data add to the previous description of the TP NLS, explore TP interaction with cellular factors, and form a basis to study the importance of TP in adenovirus infection and entry.
4. Materials and Methods
4.1. Cloning of Proteins and Fragments
The cDNAs encoding pTP, TP and other fragments were obtained from the Virapower adenovirus plasmid which was used as a template (Thermofisher, Scoresby, VIC, Australia). Fragments of the terminal protein DNA sequence were amplified and cloned using Phusion PCR master mix (NEB, Ipswich, MA, USA) and the oligonucleotides described in
Table S1. This was followed by fragment ligation to the C–terminal end of
Pontellina plumata GFP using the pMaxGFP vector (Lonza, Basel, Switzerland). The vector was first amplified with PCR (
Table S2—pMaxF, pMaxR). The linear DNA was then gel-purified before assembly. The fragments were inserted into the linear pMaxGFP using a Gibson assembly Kit (NEB, Ipswich, MA, USA), as shown in
Figure 1. Oligonucleotides were designed with the assistance of NEBbuilder website and modified using Snapgene software (v. 4.2.4, GSL Biotech; available at
https://www.snapgene.com/ ) to ensure at least 20 base-homology with the linearised pMaxGFP PCR product. All Gibson assembly reactions were carried out as recommended by the kit manufacturer, and the amplification products were verified by gel electrophoresis. After fragment assembly into the linearised pMaxGFP, the products of each reaction were transformed into DH5α
E. coli (NEB, Ipswich, MA, USA) following the manufacturer’s protocol. After streaking, growth and amplification, plasmids from the bacterial clones were purified using GeneJet plasmid miniprep (Thermofisher, Scoresby, VIC, Australia ). For mutations and deletion subfragments, the GFP–TP fragment clone was used as a template followed by PCR amplification to introduce mutations or deletions using the oligonucleotides described in
Table S1 (Mut1F/R for mutant 1, Mut2F/R for mutant 2, Mut3F/R for mutant 3 and Del1F/R for the deletion clone as shown in
Figure 1). After PCR, products were phosphorylated and ligated using T4 polynucleotide kinase and ligase (NEB, Ipswich, MA, USA) followed by transformation, selection, growth and purification as above to generate the mutated/deleted fragments. The sequence, starting at MEHFLP, was used as an alternative initiation sequence (Gene bank accession AAA92208.1). This placed Serine–580 at the alternative site of 562 (Ser562) when starting from the alternative initiation sequence. No further changes were applied.
4.2. Validation of Inserts
All fragments and mutants (Mut1, Mut2, and Del1) were sequenced downstream of the GFP C–terminus to confirm insertion of the fragments and lack of frameshift using Frag–SeqF oligo (
Table S2). Mut3 was sequenced using Frag–SeqFV2 oligo (
Table S2). Sanger sequencing was used to validate all the constructs (performed by AGFR, Melbourne, VIC, Australia).
4.3. Mammalian Expression
HeLa and 293A (Thermo Fisher, Scoresby, VIC, Australia) cells were maintained in DMEM + 10%FBS (high-glucose, pyruvate) with antibiotics. Cells were tested for mycoplasma upon their arrival and before creating frozen stocks and were free from contamination. Cells were plated onto cell culture treated µ–slide (ibidi, Martinsried, Planegg, Germany) and using 2 × 102 cells 24 h before transfection. On the following day, purified plasmids were transfected using Lipofectamine 3000/P3000 reagent (Thermofisher, Scoresby, VIC, Australia) following the recommended procedure by the manufacturer. After washing the cells, the culture medium was replaced with fresh growth medium 24 h after transfection. Forty-eight hours after transfections, the growth medium was removed and washed once with Phenol red-free L15 medium (Thermofisher, Scoresby, VIC, Australia), to facilitate growth without CO2 in the microscope chamber. This was followed by incubation with L15 medium containing 0.1 mg/mL Hoechst 33342 (Thermofisher, Scoresby, VIC, Australia) for 10 min at 37 °C. In some cases, CellMask DeepRed (Thermofisher, Scoresby, VIC, Australia) was also added to the medium to facilitate cell membrane detection and calculations. The incubation medium was then replaced again with fresh and warm L15, and cells were imaged using confocal laser microscopy with a Leica SP8 equipped with an HC PL APO CS2 63x/1.4 oil-immersion objective under 37 °C. The same settings were applied during image acquisition using LASX software (Leica, Wetzlar, Germany).
4.4. TP Cloning and Protein Expression and Purification
The ORF of TP with the TEV cleavage site close to the n–terminus was compiled using gene synthesis. In the construct, Ser-562 was replaced with cysteine residue and cloned in pET32a expression plasmid (Novagen, Darmstadt, Germany) between BamHI and EcoRI sites using directional cloning with restriction enzymes. pET32a sequencing confirmed the insertion of TP. The pET32a–TP construct was then transformed into BL21 E. coli (NEB, Ipswich, MA, USA), plated in selection LB media, and then a colony was selected for protein production. After overnight incubation in 4 × 10 mls LB culture, each culture was added to 500 mL terrific or LB broth media and incubated with orbital mixer at 37 °C, 180 rpm. The culture was monitored and then induced for 3 h with a final concentration of 1 mM Isopropyl β–D–1–thiogalactopyranoside (IPTG) at O.D.600 of 1.0–1.2. The bacteria were pelleted and then resuspended in cold lysis buffer (50 mM Tris–HCl, EDTA 10 mM, Triton–X 1% and 5% glycerol + 200 µg/mL lysozyme) and stirred using a magnetic stirrer for 20 min. The suspension was then sonicated (1 min × 3 times using 11 Wattage). Following sonication, DNAse (10 µg/mL) and MgCl2 (2 mM) were added to the suspension and incubated further for 20 min while stirring. The suspension was then spun at 10,000 g for 15 min, and the protein expression of TP was identified in the inclusion bodies. The inclusion body pellet was washed once with TritonX containing wash buffer (Tris–HCl 50 mM, TritonX 1%, 1M Urea) and with the same wash buffer lacking Triton–X twice and spun as above after each wash. A total of 30 mL of 6 M GuHCl in TrisAcetate pH 8.6 was used to resuspend the pellet and was incubated overnight at 4 °C to solubilise the protein.
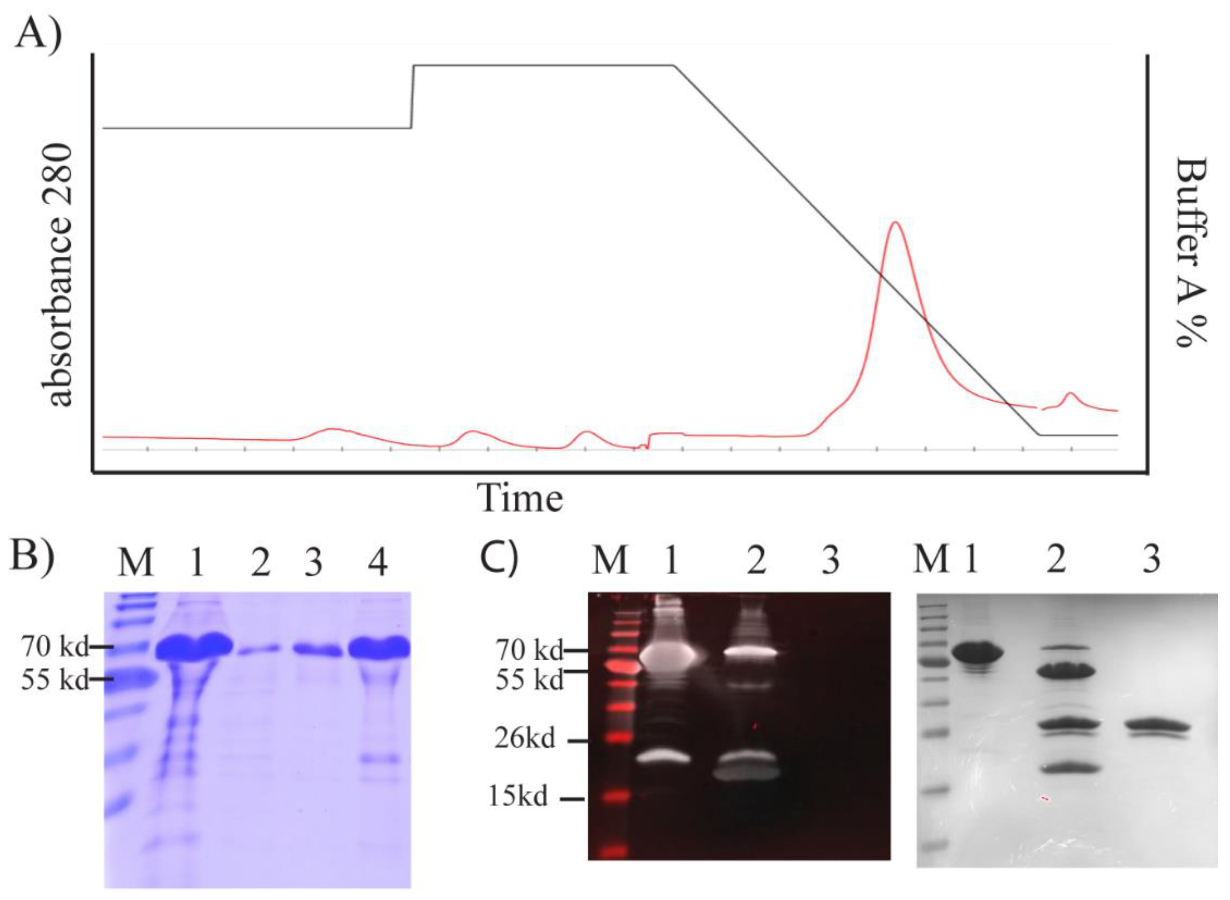
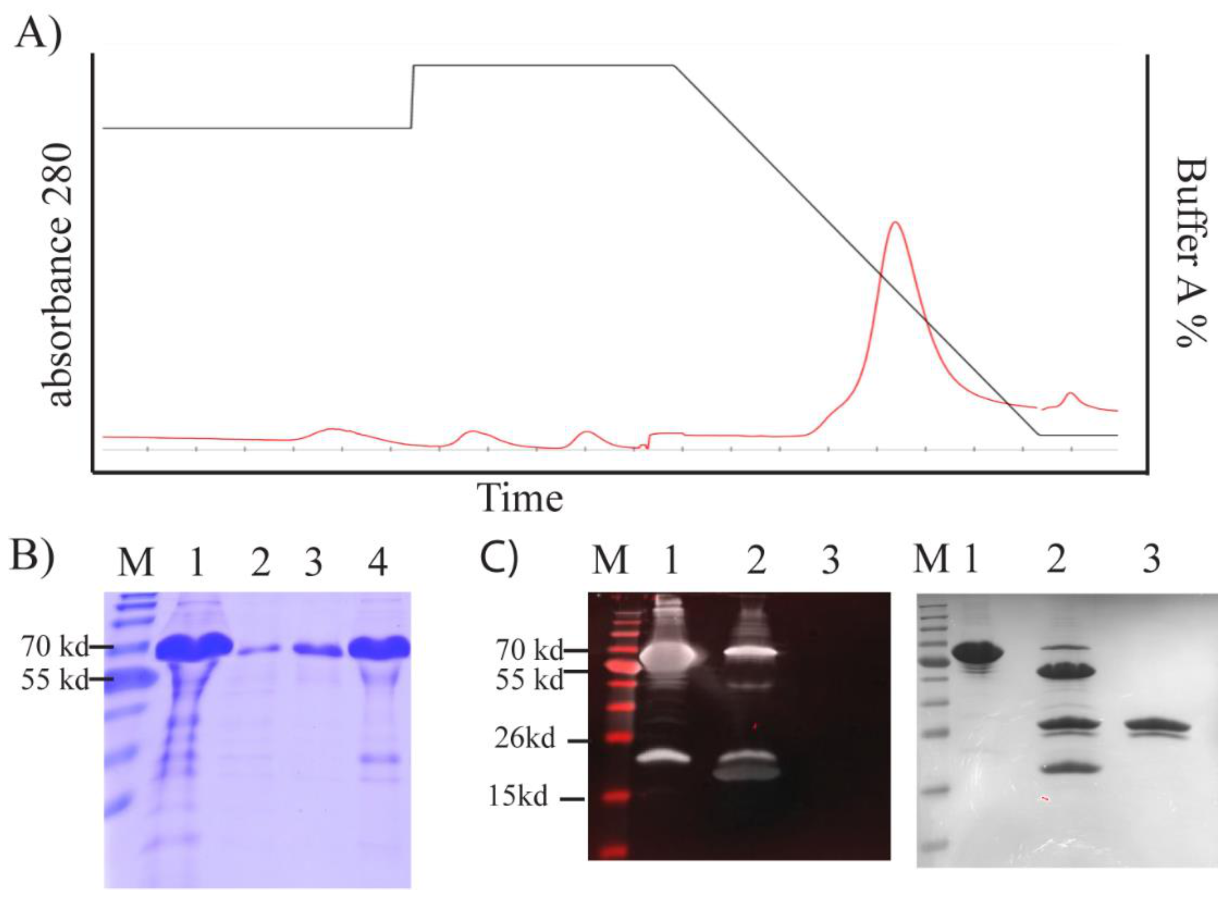
We purified TP from inclusion bodies using FPLC under denaturing conditions (
Figure 5A). This yielded high-purity TP (~91%;
Figure 5B). To obtain a high yield in a refolded form, we designed an in-house protein refolding screening assay. We selected the optimum buffer system from a group that yielded the lowest aggregation (
Figure S2). A buffer composed of 20 mM TrisAcetate, 5% glycerol, 50 mM arginine, and 50 mM glutamic acid at pH 8.4–8.6 was selected.
The TEV cleavage site was accessible, and TP was predominantly cleaved from the fusion construct after 3 h treatment with TEV enzyme (
Figure 5C). We were able to detect this cleavage by Western blotting using an antibody against the thioredoxin tag (Trx) (
Figure 5C). A band was evident above the cleaved Trx tag, but this band was barely observable with SDS–PAGE (
Figure 5C).
4.5. TP Purification and Solubility Assessment
TP–Trx containing the His-tag was purified using Histrap 5 mL column (GE healthcare). After binding, column buffer was replaced to wash buffer (20 mM TrisAcetate, 8M Urea pH 8.3) and followed by 3 column washes using the same buffer. After washing, the protein was eluted with wash buffer + 400 mM imidazole. Elution was conducted in a gradient fashion, and the protein peak was pooled.
The assessment of the refolding was performed by testing ~50 different conditions (
Figure S2), the original buffers and the additives were made in 4X concentrations initially and then diluted to 1x working concentration to contain the final concentration described in
Figure S1. A dialysis membrane of 3K was used to dialyse the TP and was lodged in an Eppendorf tube cap. The tube was cut off at the neck. The sample (50 µL of TP in urea containing buffer) was assembled inside a 50 mL tube containing 50 mL of each different dialysis buffer. The dialyses buffers were replaced once. Following overnight dialysis, the protein solutions were collected and analysed with microplate nephelometer (BMG Labtech, Offenburg, Germany) to determine the extent of aggregation and precipitation.
After determining the best dialysis buffer, the protein was then dialysed using Slide–A–Lyzer G2, MWCO 3.5KDa (Thermofisher, Scoresby, VIC, Australia) and refolded at 4 °C against 2 L dialysis buffer (20 mM TrisAcetate, 5% glycerol, 50 mM arginine, 50 mM glutamic acid pH 8.3). The dialysis buffer was replaced after 6 h, and another 2 L of the same buffer was added, and the unit remained in dialysis overnight. Next day, the protein was concentrated, aliquoted and stored at −80. TEV enzyme was expressed and purified in-house and was a gift from Professor Martin Scanlon at Monash Institute of Pharmaceutical Sciences.
To purify the thioredoxin tag to use as a control, we used TEV enzyme to cleave the TP–Trx. After, filtration was carried out with a 0.22 µM filter. Trx was separated from the other components with using FPLC SEC 70 column (Bio-Rad, Glandesville, NSW, Australia).
4.6. Western Blotting
Proteins (TP alone or TEV treated) were run in 12–4% SDS page in parallel. Half of the gel was analysed by Coomassie Blue staining, and the other half of the gel was moved onto a PVDF membrane and sandwiched inside two thick filter papers and run in Trans-Blot SD semidry electrophoresis cell (Bio-Rad, Glandesville, NSW, Australia) for 30 min at 12 V. The PVDF membrane was blocked by Odyssey® Blocking Buffer (Licor, Lincoln, NE, U.S.A.) and labelled with primary mouse Trx–tag Antibody (Assay Matrix PTY LTD) and secondary IRDye® 800 CW Goat anti-Mouse IgG (Licor, Lincoln, NE, U.S.A.) in TBST buffer (Tris-buffered-saline+ Tween 0.1% w/v) at the suggested dilutions by the manufactures. PageRuler™ Prestained NIR Protein Ladder (Thermofisher, Scoresby, VIC, Australia) was used as a molecular weight ladder.
4.7. Fluoresent Labelling of TP
TP containing the introduced cysteine was labelled with Maleimide-Alexaflour 594. The TP was first reduced with TCEP (1 mM) for 30 min. The reducing reaction was followed by spin desalting using 40K–Zeba column (Thermofisher, Scoresby, VIC, Australia) using 20 mM TrisAcetate pH 8.3 and 5% glycerol. The dye which was suspended in pure DMSO and was added to the protein under vortexing so the final concentration of the dye was 500 µM and DMSO was no more than 5% in the final solution. The mixture was incubated for two hours at room temperature and desalted twice with Zeba column as above. The dye was aliquoted and stored at −80 °C.
4.8. Cell Culture, Digitonin Permeabilisation Assay (DPA) and Microinjection
Hela cells were grown from a low passage number and were free from mycoplasma contamination after testing. For plating, cells were grown in DMEM supplemented with high glucose, pyruvate and glutamax in addition to 10% FBS (Thermofisher, Scoresby, VIC, Australia). Twenty-five thousand cells were plated onto a (22 × 22 mm) glass coverslip inside a 6-well plate for the digitonin permeabilisation assay (DPA) or in a µ–dish 35 mm (ibidi, Martinsried, Planegg, Germany) for microinjection. After plating, the cells were incubated in 37 °C, 5% CO2.
For microinjection, the cells were washed with PBS once, and DMEM was replaced with L15, phenol red-free (Thermofisher, Scoresby, VIC, Australia) and incubated in a 37 °C oven without CO
2 until the protein/dye was ready. To prepare materials for injection, 5µl TP–AF594 was mixed with 5 uL BSA–FITC [
29]. The mixture was filtered through 0.22 µM syringe filter (Merck Millipore, Bayswater, VIC, Australia) mounted on a PCR tube which was placed inside a 1.5 mL tube. The mixture was spun for 10 min at 10,000 g. In total, 2 µL of the eluted mixture was then loaded into a microneedle (Eppendorf, Macquarie Park, NSW, Australia) and the cells were injected using Eppendorf InjectMan, with settings of pc = 30 pa, pi = 50 pa, 0.2 sec. Cells were washed with L15/5% FBS once and incubated for ~20 min before imaging. Commercially available L15 media allowed the incubation of cells without the need for CO
2. Microscopy imaging of microinjection was carried out using Nikon Ti–E (Minato City, Tokyo, Japan) using the wide-field Coolsnap™ camera, and the cells were kept 37 °C during the imaging.
DPA was first optimised to determine the most appropriate digitonin concentration. Digitonin at 20 µg/µL and an incubation period of 5 min on the ice were found to be optimum conditions in our hands. The plate with coverslip-plated cells was moved on the ice and washed twice with cold transport buffer (HEPES 20 mM; potassium acetate 110 mM; sodium acetate 5 mM; magnesium acetate 2 mM; EGTA 1 mM; 1 µg/µL of leupeptin, pepstatin, and aprotinin). Cells were then incubated with 1 mL (20 µg/mL) digitonin in transport buffer for 5 min. This was followed by four washes of cold transport buffer. The coverslips were then mounted facing down on a parafilm sheet with a drop (total 50 µL) of 25 µL 2x import buffer and either 25 µL rabbit reticulocyte lysate (Promega) or 25 µL of 5 mg/mL BSA as a control. Import buffer comprised of transport buffer + energy: 1 mM ATP, 0.2 mM GTP, 5 mM creatine phosphate, 17.5 U/mL creatine phosphokinase. Finally, TP = 1 µM final concentration was used, and 1 µg of anti-DNA mouse antibody (Abcam, Cambridge, United Kingdom) was added to the final 50 µL solution.
Permeabilized cells on a drop were then incubated at 30 °C for 30 min. Cells were washed twice with cold transport buffer and once with cold PBS. Cells were then fixed with 4% paraformaldehyde (Sigma-Aldrich, North Ryde BC NSW, Australia) in PBS for 10 min at room temperature followed by two PBS–BSA 1% washes and labelling with secondary antibody (AF–488; Thermofisher, Scoresby, VIC, Australia) against anti–DNA antibody for two hours in PBS–BSA 1% buffer. Cells were then washed twice with PBS, followed by a third wash for 5 min containing 1X Hoechst counterstain (Thermofisher, Scoresby, VIC, Australia). Nuclear dye was finally washed off with PBS and coverslips were mounted on a glass slide containing a drop of antifade-gold (Thermofisher, Scoresby, VIC, Australia). Ten minutes after mounting, the edges were sealed with nail polish, and slides were either imaged the same day or were imaged the next day.
4.9. Imp α/β Protein Expression and Purification
Imp α2 and Imp β1 were expressed as GST–tagged proteins in BL21 (pREP4)
E. coli cells under native conditions and purified as previously [
30]. A GST cleaved Impβ1 protein was also prepared for use in the Impα/β1 heterodimer. Briefly, Impβ1 was bound to GST beads (GE Healthcare, North Richland Hills, TX, USA) and incubated with intracellular buffer (IB, 110 mM KCl, 5 mM NaHCO
3, 5 mM MgCl
2, 1 mM EGTA, 0.1 mM CaCl
2, 20 mM HEPES, pH 7.4), 400 µM DTT, and 10 U thrombin (Sigma-Aldrich, North Ryde BC NSW, Australia) at room temperature for 7 h. The supernatant was then added to benazamidine sepharose B beads (Pharmacia Biotech, Piscataway, NJ, USA) and incubated overnight at 4 °C. Unbound, cleaved Imp β1 protein was then collected and stored at −80 °C. Before use, the GST cleaved Imp β1, and Imp α2 were dimerised at a 1:1 ratio in IB supplemented with 1 mM DTT at a concentration of 13.6 µM for 15 min at room temperature [
31]. Protein concentrations were estimated by Coomassie staining and Bradford Protein Assay (Bio-Rad, Glandesville, NSW, Australia).
4.10. AlphaScreen Assay
AlphaScreens were performed in triplicate in opaque 384–well plates in a final volume of 25 µM as previously described [
32,
33,
34]. Briefly, 5 µL of 0.5% BSA was added to wells followed by 5 µL of TP or Trx alone (final concentration, 30 nM) and serial dilutions in 5 µL of the binding partner (importins or GST control). Plates were then incubated for 30 min to allow binding. In total, 5 µL of nickel NTA donor beads (1/250 dilution) was added to wells and incubated for 90 min, then 5 µL of GST acceptor bead (1/250 dilution) was added, and plates were incubated a further 2 h before reading on an Enspire plate reader (PerkinElmer, Waltham, Massachusetts, United States). All dilutions were in PBS, and all incubations were in the dark at room temperature. Curves were plotted using a one-phase association model in Prism (GraphPad Software, Inc., San Diego, CA, USA) to determine the dissociation constants (K
d) and the maximal signals (B
max).
4.11. Microinjection with Importins and Exportin Inhibitors
Ivermectin, leptomycin B and importazole were all purchased from Sigma. All stocks were dissolved in DMSO. Fifty thousand cells were plated using DMEM/FBS 10% as described in the microinjection section above. The cells were plated in a gridded 33 mm plate (ibidi, Martinsried, Planegg, Germany). Twenty-four hours after plating, new media containing either ivermectin (30 µM), importazole (40 µM), or leptomycin (10 ng/uL) were used with the cells after washing off the older media. Before microinjection, L15 medium containing the same concentration of the inhibitors replaced the DMEM/FBS10% to allow for CO2 free imaging. Cells were first microinjected as above using 0.2–0.3 s, 100 pa injection pressure. TP–AF594 in combination with BSA–FITC (no NLS) was used. For positive control, BSA–NLS was mixed with BSA–TexasRed. Both labelled protein solutions were filtered first before loading into the microneedle. Cells were imaged 20 min after microinjection throughout all conditions and were first incubated for 5 min with L15 + Drug + Hoechst counterstain solution (Thermofisher, Scoresby, VIC, Australia). Cells were imaged using Leica SP8 with an HC PL APO CS2 63x/1.4 oil-immersion objective (37 °C). The analysed cells were from at least 2–3 biological repeats on separate days.
4.12. Imaging, Data Collection and Analysis
Images were analysed with image-J software using a built-in script to help automate the process of analysing a large number of acquired images. This was achieved by measuring the mean green fluorescence signal in the nucleus (using the blue channel as a nuclear area mask) divided by the mean green fluorescence in the cytoplasm (N
f/C
f). N
f/C
f ratios of different fragments were then dot-plotted using GraphPad Prism, and statistical significance was determined using one-way ANOVA followed by Tukey’s multiple comparisons between all conditions within the same cell line. In some instances, Welch’s t–test was also used to compare directly between independent wells. During image acquisition, plated cells were identified, and the data were acquired to cover all transfected cells that we could find. Acquired images were either all analysed or selected randomly before analysis. Nuclear masks were produced automatically using ImageJ software; cell areas were drawn manually around every identified transfected cell using CellMask DeepRed or bright-field images as identifiers. Apoptotic cells and identifiable debris were excluded from the analysis. The acquired images were analysed using a minimum of 15 cells per condition, on average 25 cells per condition (see
Table S3 for the number of cells per each condition) and only N
f/C
f < 100 were plotted. An N
f/C
f of 100 and above signifies a complete localisation in the nucleus, and these measurements only existed in fragments where N
f/C
f was very high and had predominant nuclear localisation. The analysed cells were from 2 biological replicates (different days of transfection) using two different cell lines.
For microinjection and DPA, the Hoechst signal was used to create a nuclear mask. AF594 fluorescence intensity signal was measured inside that mask to determine nuclear localisation. Cell membranes were also outlined, and the total fluorescence intensity was measured. Finally, to calculate the cytoplasmic intensity, nuclear fluorescence was subtracted from the total cell fluorescence. Cells with nuclear positive cytoplasmic indicators were excluded as this indicates nuclear microinjection.
Data were plotted as the mean with standard deviation and analysed using either Welch’s t–test (for initial microinjection) or ANOVA with Tukey’s multiple comparisons between the conditions (in case of DPA and microinjections in the presence of inhibitors).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}