
The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration




{kind=link}
{kind=link}
Abstract
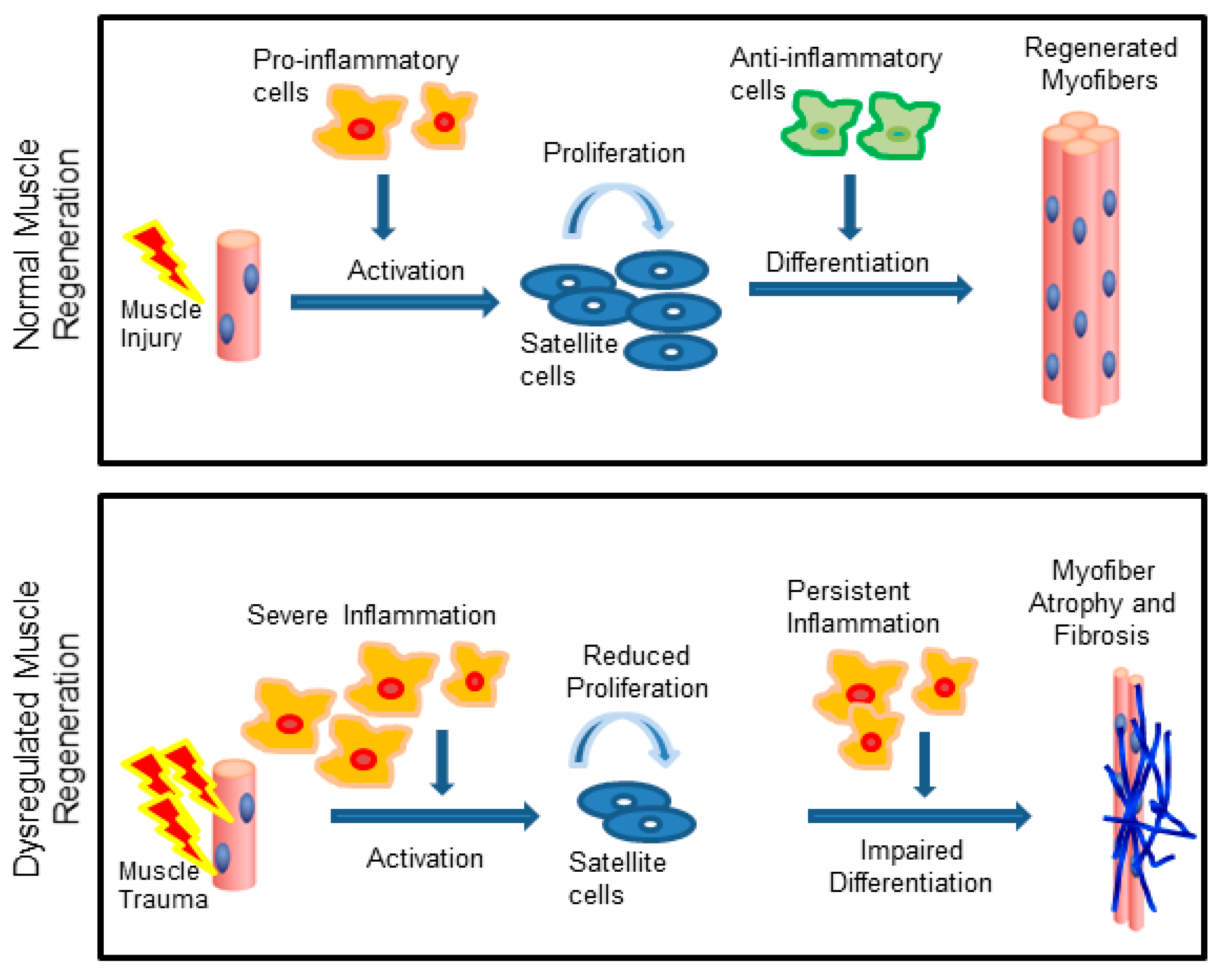
1. Introduction
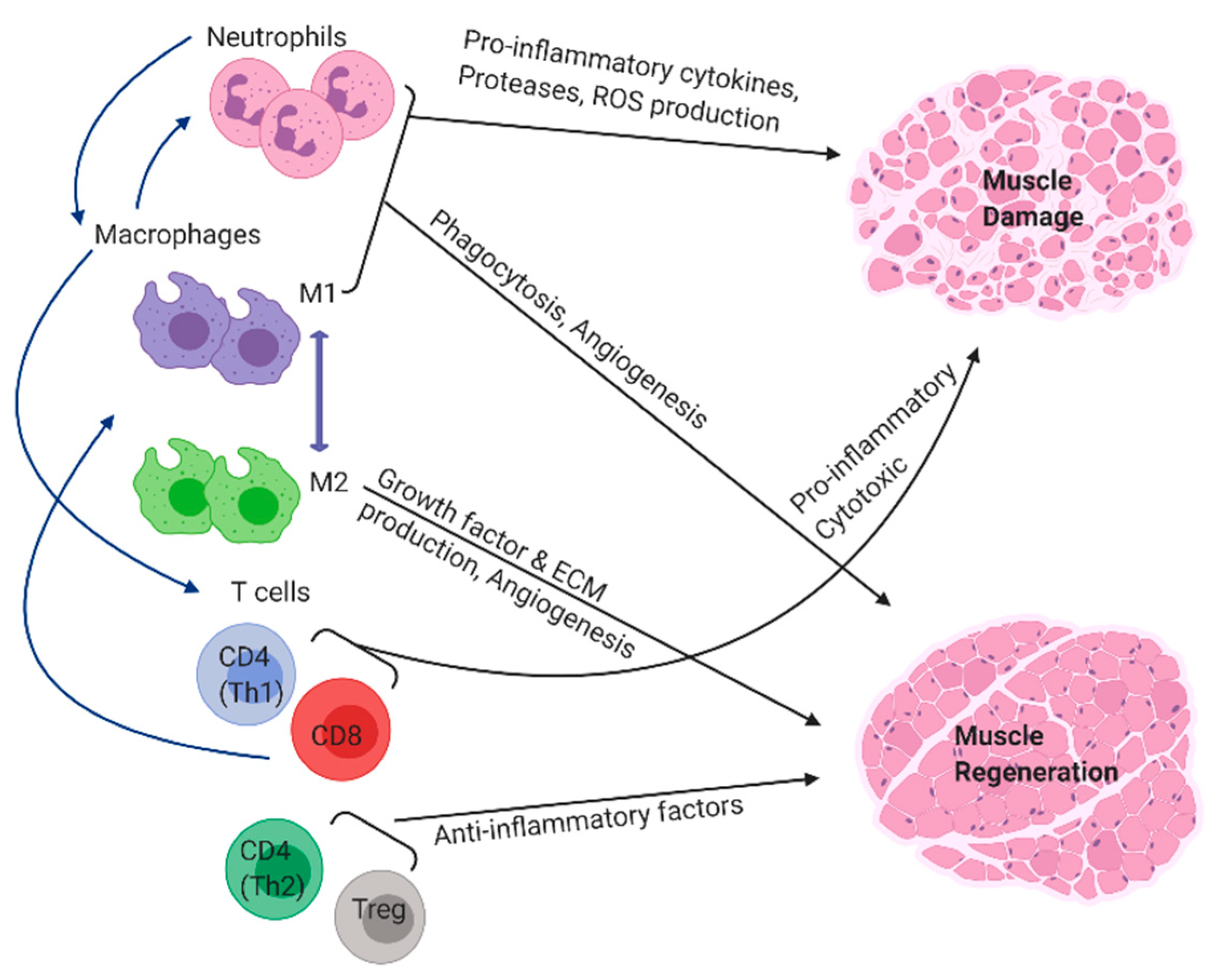
2. Innate Immune Cells Influence Muscle Regeneration
2.1. The Role of Neutrophils in Muscle Regeneration
2.2. Macrophages
2.3. Role of Macrophages in Muscle Injury
2.4. Role of Macrophages in Aged Muscle
3. Adaptive Immune Cells Mediate Muscle Regeneration
3.1. Helper and Cytotoxic T Cells
3.2. T Cell Response to Muscle Injury
3.3. T Cell Response to Exercise
3.4. Cytotoxic T Cells Mediate Continued Pathology Following Disease and Aging
3.5. Role of Treg in Acute and Chronic Muscle Injury
3.6. Role of Treg in Aged Muscle
4. Immunomodulators in Skeletal Muscle Regeneration
4.1. Immunomodulatory Properties of MSCs
4.2. Immunosuppressants as Therapies for Muscle Regeneration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef]
- Tidball, J.G. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hu, P. Skeletal muscle regeneration is modulated by inflammation. J. Orthop. Transl. 2018, 13, 25–32. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, E.; Zordan, P.; Sciorati, C.; Rovere-Querini, P.; Brunelli, S. Macrophage plasticity in skeletal muscle repair. Biomed. Res. Int. 2014, 2014, 560629. [Google Scholar] [CrossRef]
- Umansky, K.B.; Gruenbaum-Cohen, Y.; Tsoory, M.; Feldmesser, E.; Goldenberg, D.; Brenner, O.; Groner, Y. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. PLoS Genet. 2015, 11, e1005457. [Google Scholar] [CrossRef]
- Kohno, S.; Ueji, T.; Abe, T.; Nakao, R.; Hirasaka, K.; Oarada, M.; Harada-Sukeno, A.; Ohno, A.; Higashibata, A.; Mukai, R.; et al. Rantes secreted from macrophages disturbs skeletal muscle regeneration after cardiotoxin injection in Cbl-b-deficient mice. Muscle Nerve 2011, 43, 223–229. [Google Scholar] [CrossRef]
- Hurtgen, B.J.; Ward, C.L.; Garg, K.; Pollot, B.E.; Goldman, S.M.; McKinley, T.O.; Wenke, J.C.; Corona, B.T. Severe muscle trauma triggers heightened and prolonged local musculoskeletal inflammation and impairs adjacent tibia fracture healing. J. Musculoskelet. Neuronal Interact. 2016, 16, 122–134. [Google Scholar]
- Hurtgen, B.J.; Henderson, B.E.P.; Ward, C.L.; Goldman, S.M.; Garg, K.; McKinley, T.O.; Greising, S.M.; Wenke, J.C.; Corona, B.T. Impairment of early fracture healing by skeletal muscle trauma is restored by FK506. BMC Musculoskelet. Disord. 2017, 18, 253. [Google Scholar] [CrossRef]
- Brunelli, S.; Rovere-Querini, P. The immune system and the repair of skeletal muscle. Pharmacol. Res. 2008, 58, 117–121. [Google Scholar] [CrossRef]
- Butterfield, T.A.; Best, T.M.; Merrick, M.A. The dual roles of neutrophils and macrophages in inflammation: A critical balance between tissue damage and repair. J. Athl. Train 2006, 41, 457–465. [Google Scholar]
- Toumi, H.; F’Guyer, S.; Best, T.M. The role of neutrophils in injury and repair following muscle stretch. J. Anat. 2006, 208, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Pizza, F.X.; Peterson, J.M.; Baas, J.H.; Koh, T.J. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 2005, 562, 899–913. [Google Scholar] [CrossRef]
- Fielding, R.A.; Manfredi, T.J.; Ding, W.; Fiatarone, M.A.; Evans, W.J.; Cannon, J.G. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. 1993, 265, R166–R172. [Google Scholar] [CrossRef] [PubMed]
- QUINDRY, J.C.; STONE, W.L.; KING, J.; BROEDER, C.E. The effects of acute exercise on neutrophils and plasma oxidative stress. Med. Sci. Sports Exerc. 2003, 35, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Brickson, S.; Hollander, J.; Corr, D.T.; Ji, L.L.; Best, T.M. Oxidant production and immune response after stretch injury in skeletal muscle. Med. Sci. Sports Exerc. 2001, 33, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Cassatella, M.A. Neutrophil-derived proteins: Selling cytokines by the pound. Adv. Immunol. 1999, 73, 369–509. [Google Scholar] [CrossRef]
- Toumi, H.; Best, T. The inflammatory response: Friend or enemy for muscle injury? Br. J. Sports Med. 2003, 37, 284–286. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Lindborg, J.A.; Mack, M.; Zigmond, R.E. Neutrophils Are Critical for Myelin Removal in a Peripheral Nerve Injury Model of Wallerian Degeneration. J. Neurosci. 2017, 37, 10258–10277. [Google Scholar] [CrossRef] [PubMed]
- Herath, T.D.K.; Larbi, A.; Teoh, S.H.; Kirkpatrick, C.J.; Goh, B.T. Neutrophil-mediated enhancement of angiogenesis and osteogenesis in a novel triple cell co-culture model with endothelial cells and osteoblasts. J. Tissue Eng. Regen. Med. 2018, 12, e1221–e1236. [Google Scholar] [CrossRef]
- McCourt, M.; Wang, J.H.; Sookhai, S.; Redmond, H.P. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch. Surg. 1999, 134, 1325–1331, discussion 1331–1322. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.F.; Zamunér, S.R.; Zuliani, J.P.; Fernandes, C.M.; Cruz-Hofling, M.A.; Fernandes, I.; Chaves, F.; Gutiérrez, J.M. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops asper snake venom. Muscle Nerve 2003, 28, 449–459. [Google Scholar] [CrossRef]
- Dumont, N.; Bouchard, P.; Frenette, J. Neutrophil-induced skeletal muscle damage: A calculated and controlled response following hindlimb unloading and reloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1831–R1838. [Google Scholar] [CrossRef]
- Pizza, F.X.; Koh, T.J.; McGregor, S.J.; Brooks, S.V. Muscle inflammatory cells after passive stretches, isometric contractions, and lengthening contractions. J. Appl. Physiol. 2002, 92, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Frenette, J.; St-Pierre, M.; Cote, C.H.; Mylona, E.; Pizza, F.X. Muscle impairment occurs rapidly and precedes inflammatory cell accumulation after mechanical loading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R351–R357. [Google Scholar] [CrossRef]
- Brickson, S.; Ji, L.L.; Schell, K.; Olabisi, R.; St Pierre Schneider, B.; Best, T.M. M1/70 attenuates blood-borne neutrophil oxidants, activation, and myofiber damage following stretch injury. J. Appl. Physiol. 2003, 95, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Zerria, K.; Jerbi, E.; Hammami, S.; Maaroufi, A.; Boubaker, S.; Xiong, J.P.; Arnaout, M.A.; Fathallah, D.M. Recombinant integrin CD11b A-domain blocks polymorphonuclear cells recruitment and protects against skeletal muscle inflammatory injury in the rat. Immunology 2006, 119, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.X.; Tidball, J.G. Null mutation of gp91phox reduces muscle membrane lysis during muscle inflammation in mice. J. Physiol. 2003, 553, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.X.; Lusis, A.J.; Tidball, J.G. Null mutation of myeloperoxidase in mice prevents mechanical activation of neutrophil lysis of muscle cell membranes in vitro and in vivo. J. Physiol. 2005, 565, 403–413. [Google Scholar] [CrossRef]
- Kawanishi, N.; Mizokami, T.; Niihara, H.; Yada, K.; Suzuki, K. Neutrophil Depletion Attenuates Muscle Injury after Exhaustive Exercise. Med. Sci. Sports Exerc. 2016, 48, 1917–1924. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Sokolow, S.; Lee, J.J.; Myung, K.H.; Villalta, S.A.; Tidball, J.G. Major basic protein-1 promotes fibrosis of dystrophic muscle and attenuates the cellular immune response in muscular dystrophy. Hum. Mol. Genet. 2008, 17, 2280–2292. [Google Scholar] [CrossRef] [PubMed]
- Crinnion, J.N.; Homer-Vanniasinkam, S.; Hatton, R.; Parkin, S.M.; Gough, M.J. Role of neutrophil depletion and elastase inhibition in modifying skeletal muscle reperfusion injury. Cardiovasc. Surg. 1994, 2, 749–753. [Google Scholar] [PubMed]
- Kyriakides, C.; Austen, W., Jr.; Wang, Y.; Favuzza, J.; Kobzik, L.; Moore, F.D., Jr.; Hechtman, H.B. Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am. J. Physiol. 1999, 277, C1263–C1268. [Google Scholar] [CrossRef]
- Walden, D.L.; McCutchan, H.J.; Enquist, E.G.; Schwappach, J.R.; Shanley, P.F.; Reiss, O.K.; Terada, L.S.; Leff, J.A.; Repine, J.E. Neutrophils accumulate and contribute to skeletal muscle dysfunction after ischemia-reperfusion. Am. J. Physiol. 1990, 259, H1809–H1812. [Google Scholar] [CrossRef]
- Arecco, N.; Clarke, C.J.; Jones, F.K.; Simpson, D.M.; Mason, D.; Beynon, R.J.; Pisconti, A. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci. Rep. 2016, 6, 24708. [Google Scholar] [CrossRef]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef]
- Grounds, M.D.; Torrisi, J. Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 676–682. [Google Scholar] [CrossRef]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Ghaly, A.; Marsh, D.R. Aging-associated oxidative stress modulates the acute inflammatory response in skeletal muscle after contusion injury. Exp. Gerontol. 2010, 45, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Peake, J.; Della Gatta, P.; Cameron-Smith, D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1485–R1495. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D. Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain, age, and sex of host mice. J. Pathol. 1987, 153, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Juban, G.; Chazaud, B. Metabolic regulation of macrophages during tissue repair: Insights from skeletal muscle regeneration. FEBS Lett. 2017, 591, 3007–3021. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Melton, D.W.; Porter, L.; Sarwar, Z.U.; McManus, L.M.; Shireman, P.K. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am. J. Pathol. 2014, 184, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Jia, T.; Hohl, T.M.; Pamer, E.G. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008, 26, 421–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, Y.; Zhao, L.; Zeng, Z.; Xiao, W.; Chen, P. Macrophage depletion impairs skeletal muscle regeneration: The roles of regulatory factors for muscle regeneration. Cell Biol. Int. 2017, 41, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Brigitte, M.; Schilte, C.; Plonquet, A.; Baba-Amer, Y.; Henri, A.; Charlier, C.; Tajbakhsh, S.; Albert, M.; Gherardi, R.K.; Chrétien, F. Muscle resident macrophages control the immune cell reaction in a mouse model of notexin-induced myoinjury. Arthritis Rheumatol. 2010, 62, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Mass, E. Delineating the origins, developmental programs and homeostatic functions of tissue-resident macrophages. Int. Immunol. 2018, 30, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef]
- Tonkin, J.; Temmerman, L.; Sampson, R.D.; Gallego-Colon, E.; Barberi, L.; Bilbao, D.; Schneider, M.D.; Musarò, A.; Rosenthal, N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol. Ther. 2015, 23, 1189–1200. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Pillon, N.J.; Bilan, P.J.; Fink, L.N.; Klip, A. Cross-talk between skeletal muscle and immune cells: Muscle-derived mediators and metabolic implications. Am. J. Physiol. -Endocrinol. Metab. 2012, 304, E453–E465. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, J.-B.; He, Y.-L.; Peng, J.-J.; Zhang, X.-H.; Chen, C.-Q.; Li, W.; Cai, S.-R. Tumor-associated macrophages promote angiogenesis and lymphangiogenesis of gastric cancer. J. Surg. Oncol. 2012, 106, 462–468. [Google Scholar] [CrossRef]
- Bosurgi, L.; Manfredi, A.A.; Rovere-Querini, P. Macrophages in injured skeletal muscle: A perpetuum mobile causing and limiting fibrosis, prompting or restricting resolution and regeneration. Front. Immunol. 2011, 2, 62. [Google Scholar] [CrossRef]
- Zhao, W.; Lu, H.; Wang, X.; Ransohoff, R.M.; Zhou, L. CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 380–393. [Google Scholar] [CrossRef]
- Summan, M.; Warren, G.L.; Mercer, R.R.; Chapman, R.; Hulderman, T.; Van Rooijen, N.; Simeonova, P.P. Macrophages and skeletal muscle regeneration: A clodronate-containing liposome depletion study. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2006, 290, R1488–R1495. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Wehling-Henricks, M. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol. 2007, 578, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Bencze, M.; Negroni, E.; Vallese, D.; Yacoub-Youssef, H.; Chaouch, S.; Wolff, A.; Aamiri, A.; Di Santo, J.P.; Chazaud, B.; Butler-Browne, G.; et al. Proinflammatory Macrophages Enhance the Regenerative Capacity of Human Myoblasts by Modifying Their Kinetics of Proliferation and Differentiation. Mol. Ther. 2012, 20, 2168–2179. [Google Scholar] [CrossRef]
- Sonnet, C.; Lafuste, P.; Arnold, L.; Brigitte, M.; Poron, F.; Authier, F.; Chrétien, F.; Gherardi, R.K.; Chazaud, B. Human macrophages rescue myoblasts and myotubes from apoptosis through a set of adhesion molecular systems. J. Cell Sci. 2006, 119, 2497–2507. [Google Scholar] [CrossRef]
- Chazaud, B.; Sonnet, C.; Lafuste, P.; Bassez, G.; Rimaniol, A.-C.; Poron, F.; Authier, F.-J.; Dreyfus, P.A.; Gherardi, R.K. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 2003, 163, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Cuvellier, S.; Magnan, M.; Mounier, R.; Chazaud, B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013, 280, 4118–4130. [Google Scholar] [CrossRef]
- Arnold, L.; Perrin, H.; de Chanville, C.B.; Saclier, M.; Hermand, P.; Poupel, L.; Guyon, E.; Licata, F.; Carpentier, W.; Vilar, J.; et al. CX3CR1 deficiency promotes muscle repair and regeneration by enhancing macrophage ApoE production. Nat. Commun. 2015, 6, 8972. [Google Scholar] [CrossRef]
- Warren, G.L.; Hulderman, T.; Mishra, D.; Gao, X.; Millecchia, L.; O’Farrell, L.; Kuziel, W.A.; Simeonova, P.P. Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J. 2005, 19, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of Ccl2, Cx3cr1 and Ccr5 abrogates Ly6chi and Ly6clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, Z.; Qu, C.; Cui, W.; Wang, X.; Du, J. CD8 T cells are involved in skeletal muscle regeneration through facilitating MCP-1 secretion and Gr1high macrophage infiltration. J. Immunol. 2014, 193, 5149–5160. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.O.; McHale, M.J.; Wells, J.T.; Ochoa, O.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Regulation of skeletal muscle regeneration by CCR2-activating chemokines is directly related to macrophage recruitment. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R832–R842. [Google Scholar] [CrossRef] [PubMed]
- Melton, D.W.; Roberts, A.C.; Wang, H.; Sarwar, Z.; Wetzel, M.D.; Wells, J.T.; Porter, L.; Berton, M.T.; McManus, L.M.; Shireman, P.K. Absence of CCR2 results in an inflammaging environment in young mice with age-independent impairments in muscle regeneration. J. Leukoc. Biol. 2016, 100, 1011–1025. [Google Scholar] [CrossRef]
- Ochoa, O.; Sun, D.; Reyes-Reyna, S.M.; Waite, L.L.; Michalek, J.E.; McManus, L.M.; Shireman, P.K. Delayed angiogenesis and VEGF production in CCR2−/− mice during impaired skeletal muscle regeneration. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2007, 293, R651–R661. [Google Scholar] [CrossRef]
- Contreras-Shannon, V.; Ochoa, O.; Reyes-Reyna, S.M.; Sun, D.; Michalek, J.E.; Kuziel, W.A.; McManus, L.M.; Shireman, P.K. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am. J. Physiol.-Cell. Physiol. 2007, 292, C953–C967. [Google Scholar] [CrossRef]
- Shireman, P.K.; Contreras-Shannon, V.; Ochoa, O.; Karia, B.P.; Michalek, J.E.; McManus, L.M. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol. 2007, 81, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Huang, D.; Saederup, N.; Charo, I.F.; Ransohoff, R.M.; Zhou, L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Huang, D.; Ransohoff, R.M.; Zhou, L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3344–3355. [Google Scholar] [CrossRef]
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Novak, M.L.; Weinheimer-Haus, E.M.; Koh, T.J. Macrophage activation and skeletal muscle healing following traumatic injury. J. Pathol. 2014, 232, 344–355. [Google Scholar] [CrossRef]
- Rybalko, V.; Hsieh, P.-L.; Merscham-Banda, M.; Suggs, L.J.; Farrar, R.P. The Development of Macrophage-Mediated Cell Therapy to Improve Skeletal Muscle Function after Injury. PLoS ONE 2015, 10, e0145550. [Google Scholar] [CrossRef] [PubMed]
- Filippin, L.I.; Moreira, A.J.; Marroni, N.P.; Xavier, R.M. Nitric oxide and repair of skeletal muscle injury. Nitric Oxide 2009, 21, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Yamamuro, A.; Maeda, S. Nitric oxide at a low concentration protects murine macrophage RAW264 cells against nitric oxide-induced death via cGMP signaling pathway. Br. J. Pharm. 2003, 139, 28–34. [Google Scholar] [CrossRef]
- Wink, D.A.; Hanbauer, I.; Laval, F.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. Nitric Oxide Protects against the Cytotoxic Effects of Reactive Oxygen Species. Ann. N. Y. Acad. Sci. 1994, 738, 265–278. [Google Scholar] [CrossRef]
- Fujii, Y.; Guo, Y.; Hussain, S.N.A. Regulation of nitric oxide production in response to skeletal muscle activation. J. Appl. Physiol. 1998, 85, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191. [Google Scholar] [CrossRef]
- Filippin, L.I.; Cuevas, M.J.; Lima, E.; Marroni, N.P.; Gonzalez-Gallego, J.; Xavier, R.M. Nitric oxide regulates the repair of injured skeletal muscle. Nitric Oxide 2011, 24, 43–49. [Google Scholar] [CrossRef]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.C.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef]
- Deng, B.; Wehling-Henricks, M.; Villalta, S.A.; Wang, Y.; Tidball, J.G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 2012, 189, 3669–3680. [Google Scholar] [CrossRef]
- Walton, R.G.; Kosmac, K.; Mula, J.; Fry, C.S.; Peck, B.D.; Groshong, J.S.; Finlin, B.S.; Zhu, B.; Kern, P.A.; Peterson, C.A. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Sci. Rep. 2019, 9, 969. [Google Scholar] [CrossRef]
- Hachim, D.; LoPresti, S.T.; Yates, C.C.; Brown, B.N. Shifts in macrophage phenotype at the biomaterial interface via IL-4 eluting coatings are associated with improved implant integration. Biomaterials 2017, 112, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.E.; Kim, D.S.; Chung, H.C.; Shing, B.; Moon, K.H.; George, S.K.; Kim, M.W.; Atala, Z.; Kim, J.H.; Ko, I.K.; et al. Controlled Delivery of Stem Cell-Derived Trophic Factors Accelerates Kidney Repair After Renal Ischemia-Reperfusion Injury in Rats. Stem. Cells Transl. Med. 2019, 8, 959–970. [Google Scholar] [CrossRef]
- Sok, M.C.P.; Baker, N.; McClain, C.; Lim, H.S.; Turner, T.; Hymel, L.; Ogle, M.; Olingy, C.; Palacios, J.I.; Garcia, J.R.; et al. Dual delivery of IL-10 and AT-RvD1 from PEG hydrogels polarize immune cells towards pro-regenerative phenotypes. Biomaterials 2021, 268, 120475. [Google Scholar] [CrossRef]
- Potas, J.R.; Haque, F.; Maclean, F.L.; Nisbet, D.R. Interleukin-10 conjugated electrospun polycaprolactone (PCL) nanofibre scaffolds for promoting alternatively activated (M2) macrophages around the peripheral nerve in vivo. J. Immunol. Methods 2015, 420, 38–49. [Google Scholar] [CrossRef]
- Garg, K.; Pullen, N.A.; Oskeritzian, C.A.; Ryan, J.J.; Bowlin, G.L. Macrophage functional polarization (M1/M2) in response to varying fiber and pore dimensions of electrospun scaffolds. Biomaterials 2013, 34, 4439–4451. [Google Scholar] [CrossRef]
- Hsieh, P.-L.; Rybalko, V.; Baker, A.B.; Suggs, L.J.; Farrar, R.P. Recruitment and therapeutic application of macrophages in skeletal muscles after hind limb ischemia. J. Vasc. Surg. 2018, 67, 1908–1920.e1901. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rinaldi, C.; Deng, B.; Liu, G.; Fedor, B.; Tidball, J.G. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum. Mol. Genet. 2011, 20, 790–805. [Google Scholar] [CrossRef]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef] [PubMed]
- Segawa, M.; Fukada, S.-I.; Yamamoto, Y.; Yahagi, H.; Kanematsu, M.; Sato, M.; Ito, T.; Uezumi, A.; Hayashi, S.i.; Miyagoe-Suzuki, Y.; et al. Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp. Cell Res. 2008, 314, 3232–3244. [Google Scholar] [CrossRef] [PubMed]
- Greising, S.M.; Rivera, J.C.; Goldman, S.M.; Watts, A.; Aguilar, C.A.; Corona, B.T. Unwavering Pathobiology of Volumetric Muscle Loss Injury. Sci. Rep. 2017, 7, 13179. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, C.A.; Greising, S.M.; Watts, A.; Goldman, S.M.; Peragallo, C.; Zook, C.; Larouche, J.; Corona, B.T. Multiscale analysis of a regenerative therapy for treatment of volumetric muscle loss injury. Cell Death. Discov. 2018, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Ward, C.L.; Rathbone, C.R.; Corona, B.T. Transplantation of devitalized muscle scaffolds is insufficient for appreciable de novo muscle fiber regeneration after volumetric muscle loss injury. Cell Tissue. Res. 2014, 358, 857–873. [Google Scholar] [CrossRef]
- Garg, K.; Ward, C.L.; Corona, B.T. Asynchronous inflammation and myogenic cell migration limit muscle tissue regeneration mediated by a cellular scaffolds. Inflamm Cel. Signal. 2014, 1. [Google Scholar] [CrossRef]
- Roth, S.M.; Metter, E.J.; Ling, S.; Ferrucci, L. Inflammatory factors in age-related muscle wasting. Curr. Opin. Rheumatol. 2006, 18, 625–630. [Google Scholar] [CrossRef]
- Hepple, R.T.; Rice, C.L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2016, 594, 1965–1978. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef]
- Wang, Y.; Wehling-Henricks, M.; Welc, S.S.; Fisher, A.L.; Zuo, Q.; Tidball, J.G. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 2019, 33, 1415–1427. [Google Scholar] [CrossRef]
- Przybyla, B.; Gurley, C.; Harvey, J.F.; Bearden, E.; Kortebein, P.; Evans, W.J.; Sullivan, D.H.; Peterson, C.A.; Dennis, R.A. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp. Gerontol. 2006, 41, 320–327. [Google Scholar] [CrossRef]
- Cui, C.-Y.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18, e13032. [Google Scholar] [CrossRef]
- Tam, C.S.; Sparks, L.M.; Johannsen, D.L.; Covington, J.D.; Church, T.S.; Ravussin, E. Low Macrophage Accumulation in Skeletal Muscle of Obese Type 2 Diabetics and Elderly Subjects. Obesity 2012, 20, 1530–1533. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wehling-Henricks, M.; Samengo, G.; Tidball, J.G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 2015, 14, 678–688. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Jordan, M.C.; Gotoh, T.; Grody, W.W.; Roos, K.P.; Tidball, J.G. Arginine Metabolism by Macrophages Promotes Cardiac and Muscle Fibrosis in mdx Muscular Dystrophy. PLoS ONE 2010, 5, e10763. [Google Scholar] [CrossRef]
- Sicari, B.M.; Johnson, S.A.; Siu, B.F.; Crapo, P.M.; Daly, K.A.; Jiang, H.; Medberry, C.J.; Tottey, S.; Turner, N.J.; Badylak, S.F. The effect of source animal age upon the in vivo remodeling characteristics of an extracellular matrix scaffold. Biomaterials 2012, 33, 5524–5533. [Google Scholar] [CrossRef] [PubMed]
- Cano, R.L.E.; Lopera, H.D.E. Introduction to T and B lymphocytes. In Autoimmunity from Bench to Bedside [Internet]; Chapter 5; Anaya, J.M., Shoenfeld, Y., Rojas-Villarraga, A., Levy, R.A., Cervera, R., Eds.; El Rosario University Press: Bogota, Colombia, 18 July 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459471/ (accessed on 21 March 2021).
- Sun, W.; Wei, F.Q.; Li, W.J.; Wei, J.W.; Zhong, H.; Wen, Y.H.; Lei, W.B.; Chen, L.; Li, H.; Lin, H.Q.; et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 2017, 117, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Fanelli, G.; Tan, N.; Nova-Lamperti, E.; McGregor, R.; Lechler, R.I.; Lombardi, G.; Scottà, C. Expanded Regulatory T Cells Induce Alternatively Activated Monocytes with a Reduced Capacity to Expand T Helper-17 Cells. Front. Immunol. 2018, 9, 1625. [Google Scholar] [CrossRef]
- Schmidt, A.; Zhang, X.M.; Joshi, R.N.; Iqbal, S.; Wahlund, C.; Gabrielsson, S.; Harris, R.A.; Tegnér, J. Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-β. Immunol. Cell Biol. 2016, 94, 747–762. [Google Scholar] [CrossRef]
- Shephard, R.J.; Rhind, S.; Shek, P.N. Exercise and the immune system. Natural killer cells, interleukins and related responses. Sports Med. 1994, 18, 340–369. [Google Scholar] [CrossRef]
- Marshall-Gradisnik, S.; Weatherby, R.; Deakin, G.; Coutts, R.; Meir, R.; Connellan, P.; Stevenson, L.; Rogerson, S. Natural killer cell activity following 6 weeks of strength training in healthy young males with/without testosterone enanthate administration. J. Exerc. Sci. Fit. 2009, 6, 106–114. [Google Scholar]
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Investig. 1996, 97, 2905–2910. [Google Scholar] [CrossRef]
- Fu, X.; Xiao, J.; Wei, Y.; Li, S.; Liu, Y.; Yin, J.; Sun, K.; Sun, H.; Wang, H.; Zhang, Z.; et al. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015, 25, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Al-Shanti, N.; Durcan, P.; Al-Dabbagh, S.; Dimchev, G.A.; Stewart, C.E. Activated lymphocytes secretome inhibits differentiation and induces proliferation of C2C12 myoblasts. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 33, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Nguyen, M.H.; Fantuzzi, G.; Koh, T.J. Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am. J. Physiol. Cell. Physiol. 2008, 294, C1183–C1191. [Google Scholar] [CrossRef]
- Hurtgen, B.J.; Ward, C.L.; Leopold Wager, C.M.; Garg, K.; Goldman, S.M.; Henderson, B.E.P.; McKinley, T.O.; Greising, S.M.; Wenke, J.C.; Corona, B.T. Autologous minced muscle grafts improve endogenous fracture healing and muscle strength after musculoskeletal trauma. Physiol. Rep. 2017, 5, e13362. [Google Scholar] [CrossRef] [PubMed]
- Sadtler, K.; Estrellas, K.; Allen, B.W.; Wolf, M.T.; Fan, H.; Tam, A.J.; Patel, C.H.; Luber, B.S.; Wang, H.; Wagner, K.R.; et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 2016, 352, 366–370. [Google Scholar] [CrossRef]
- Kwee, B.J.; Budina, E.; Najibi, A.J.; Mooney, D.J. CD4 T-cells regulate angiogenesis and myogenesis. Biomaterials 2018, 178, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Sangregorio, R.; Sartirana, C.; Moschin, A.L.; Battaglia, M.; Orban, P.C.; Roncarolo, M.-G. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor beta, but not interleukin 10, and are distinct from type 1 T regulatory cells. J. Exp. Med. 2002, 196, 1335–1346. [Google Scholar] [CrossRef]
- Dumke, B.R.; Lees, S.J. Age-related impairment of T cell-induced skeletal muscle precursor cell function. Am. J. Physiol. Cell Physiol. 2011, 300, C1226–C1233. [Google Scholar] [CrossRef] [PubMed]
- Malm, C.; Nyberg, P.; Engstrom, M.; Sjodin, B.; Lenkei, R.; Ekblom, B.; Lundberg, I. Immunological changes in human skeletal muscle and blood after eccentric exercise and multiple biopsies. J. Physiol. 2000, 529 Pt 1, 243–262. [Google Scholar] [CrossRef]
- Malm, C.; Sjodin, T.L.; Sjoberg, B.; Lenkei, R.; Renstrom, P.; Lundberg, I.E.; Ekblom, B. Leukocytes, cytokines, growth factors and hormones in human skeletal muscle and blood after uphill or downhill running. J. Physiol. 2004, 556, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- Marklund, P.; Mattsson, C.M.; Wahlin-Larsson, B.; Ponsot, E.; Lindvall, B.; Lindvall, L.; Ekblom, B.; Kadi, F. Extensive inflammatory cell infiltration in human skeletal muscle in response to an ultraendurance exercise bout in experienced athletes. J. Appl. Physiol. 2013, 114, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Deyhle, M.R.; Gier, A.M.; Evans, K.C.; Eggett, D.L.; Nelson, W.B.; Parcell, A.C.; Hyldahl, R.D. Skeletal Muscle Inflammation Following Repeated Bouts of Lengthening Contractions in Humans. Front. Physiol. 2015, 6, 424. [Google Scholar] [CrossRef]
- Hyldahl, R.D.; Chen, T.C.; Nosaka, K. Mechanisms and Mediators of the Skeletal Muscle Repeated Bout Effect. Exerc. Sport Sci. Rev. 2017, 45, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Deyhle, M.R.; Hyldahl, R.D. The Role of T Lymphocytes in Skeletal Muscle Repair from Traumatic and Contraction-Induced Injury. Front. Physiol. 2018, 9, 768. [Google Scholar] [CrossRef] [PubMed]
- Ronsen, O.; Pedersen, B.K.; Oritsland, T.R.; Bahr, R.; Kjeldsen-Kragh, J. Leukocyte counts and lymphocyte responsiveness associated with repeated bouts of strenuous endurance exercise. J. Appl. Physiol. 2001, 91, 425–434. [Google Scholar] [CrossRef]
- Gleeson, M.; Bishop, N.C. The T cell and NK cell immune response to exercise. Ann. Transpl. 2005, 10, 43–48. [Google Scholar]
- Campbell, J.P.; Turner, J.E. Debunking the Myth of Exercise-Induced Immune Suppression: Redefining the Impact of Exercise on Immunological Health across the Lifespan. Front. Immunol. 2018, 9, 648. [Google Scholar] [CrossRef]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef]
- Garritson, J.; Krynski, L.; Haverbeck, L.; Haughian, J.M.; Pullen, N.A.; Hayward, R. Physical activity delays accumulation of immunosuppressive myeloid-derived suppressor cells. PLoS ONE 2020, 15, e0234548. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Walsh, C.M.; Dorshkind, K.A.; Rodriguez, E.M.; Tidball, J.G. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Investig. 1997, 99, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Madaro, L.; Bouche, M. From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: The role of lymphocytes. Biomed. Res. Int. 2014, 2014, 438675. [Google Scholar] [CrossRef]
- Spencer, M.J.; Montecino-Rodriguez, E.; Dorshkind, K.; Tidball, J.G. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 2001, 98, 235–243. [Google Scholar] [CrossRef]
- Emslie-Smith, A.M.; Arahata, K.; Engel, A.G. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell-mediated cytotoxicity in myopathies. Hum. Pathol. 1989, 20, 224–231. [Google Scholar] [CrossRef]
- Choi, J.H.; Park, Y.E.; Kim, S.I.; Kim, J.I.; Lee, C.H.; Park, K.H.; Kim, D.S. Differential immunohistological features of inflammatory myopathies and dysferlinopathy. J. Korean Med. Sci. 2009, 24, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Sitzia, C.; Navarro, C.; D’Antona, G.; Belicchi, M.; Parolini, D.; Del Fraro, G.; Razini, P.; Bottinelli, R.; Meregalli, M.; et al. Absence of T and B lymphocytes modulates dystrophic features in dysferlin deficient animal model. Exp. Cell Res. 2012, 318, 1160–1174. [Google Scholar] [CrossRef]
- Farini, A.; Meregalli, M.; Belicchi, M.; Battistelli, M.; Parolini, D.; D’Antona, G.; Gavina, M.; Ottoboni, L.; Constantin, G.; Bottinelli, R.; et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007, 213, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Ouisse, L.-H.; Remy, S.; Lafoux, A.; Larcher, T.; Tesson, L.; Chenouard, V.; Guillonneau, C.; Brusselle, L.; Vimond, N.; Rouger, K.; et al. Immunological characterization of a rat model of Duchenne’s disease and demonstration of improved muscle strength after anti-CD45RC antibody treatment. Front. Immunol. 2019, 10, 2131. [Google Scholar] [CrossRef]
- Madaro, L.; Pelle, A.; Nicoletti, C.; Crupi, A.; Marrocco, V.; Bossi, G.; Soddu, S.; Bouche, M. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS ONE 2012, 7, e31515. [Google Scholar] [CrossRef]
- Ieronimakis, N.; Pantoja, M.; Hays, A.L.; Dosey, T.L.; Qi, J.; Fischer, K.A.; Hoofnagle, A.N.; Sadilek, M.; Chamberlain, J.S.; Ruohola-Baker, H.; et al. Increased sphingosine-1-phosphate improves muscle regeneration in acutely injured mdx mice. Skelet Muscle 2013, 3, 20. [Google Scholar] [CrossRef]
- Jin, Y.; Knudsen, E.; Wang, L.; Bryceson, Y.; Damaj, B.; Gessani, S.; Maghazachi, A.A. Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood 2003, 101, 4909–4915. [Google Scholar] [CrossRef]
- Eghtesad, S.; Jhunjhunwala, S.; Little, S.R.; Clemens, P.R. Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol. Med. 2011, 17, 917–924. [Google Scholar] [CrossRef]
- Morrison, J.; Palmer, D.B.; Cobbold, S.; Partridge, T.; Bou-Gharios, G. Effects of T-lymphocyte depletion on muscle fibrosis in the mdx mouse. Am. J. Pathol. 2005, 166, 1701–1710. [Google Scholar] [CrossRef]
- Sugiura, T.; Murakawa, Y.; Nagai, A.; Kondo, M.; Kobayashi, S. Fas and Fas ligand interaction induces apoptosis in inflammatory myopathies: CD4+ T cells cause muscle cell injury directly in polymyositis. Arthritis Rheumatol. 1999, 42, 291–298. [Google Scholar] [CrossRef]
- Mizuno, K.; Yachie, A.; Nagaoki, S.; Wada, H.; Okada, K.; Kawachi, M.; Toma, T.; Konno, A.; Ohta, K.; Kasahara, Y.; et al. Oligoclonal expansion of circulating and tissue-infiltrating CD8+ T cells with killer/effector phenotypes in juvenile dermatomyositis syndrome. Clin. Exp. Immunol. 2004, 137, 187–194. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Koarada, S.; Tada, Y.; Ushiyama, O.; Morito, F.; Suzuki, N.; Ohta, A.; Horiuchi, T.; Miyake, K.; Nagasawa, K. Difference in B cell activation between dermatomyositis and polymyositis: Analysis of the expression of RP105 on peripheral blood B cells. Ann. Rheum. Dis. 2001, 60, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Green, J.A.; Moltedo, B.; Arvey, A.; Hemmers, S.; Yuan, S.; Treuting, P.M.; Rudensky, A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 2015, 162, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.P.; Sheng, D.Z.; Sugimoto, K.; Gonzalez-Rajal, A.; Nakagawa, S.; Hesselson, D.; Kikuchi, K. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 2017, 43, 659–672.e655. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar] [CrossRef] [PubMed]
- Thornton, A.M.; Korty, P.E.; Kim, Y.C.; Martens, C.; Shevach, E.M. Helios expression defines a phenotypically distinct population of Treg cells. J. Immunol. 2018, 200, 116–119. [Google Scholar]
- Yadav, M.; Louvet, C.; Davini, D.; Gardner, J.M.; Martinez-Llordella, M.; Bailey-Bucktrout, S.; Anthony, B.A.; Sverdrup, F.M.; Head, R.; Kuster, D.J.; et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 2012, 209, 1713–S1719. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [PubMed]
- DiSpirito, J.R.; Zemmour, D.; Ramanan, D.; Cho, J.; Zilionis, R.; Klein, A.M.; Benoist, C.; Mathis, D. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci. Immunol. 2018, 3, eaat5861. [Google Scholar] [CrossRef]
- Jin, R.M.; Warunek, J.; Wohlfert, E.A. Therapeutic administration of IL-10 and amphiregulin alleviates chronic skeletal muscle inflammation and damage induced by infection. Immunohorizons 2018, 2, 142–154. [Google Scholar] [CrossRef]
- Beyer, M.; Thabet, Y.; Müller, R.-U.; Sadlon, T.; Classen, S.; Lahl, K.; Basu, S.; Zhou, X.; Bailey-Bucktrout, S.L.; Krebs, W.; et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat. Immunol. 2011, 12, 898. [Google Scholar] [CrossRef]
- Alvarez, J.D.; Yasui, D.H.; Niida, H.; Joh, T.; Loh, D.Y.; Kohwi-Shigematsu, T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 2000, 14, 521–535. [Google Scholar] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug. Discov. 2008, 7, 827. [Google Scholar] [CrossRef]
- Taams, L.S.; van Amelsfort, J.M.R.; Tiemessen, M.M.; Jacobs, K.M.G.; de Jong, E.C.; Akbar, A.N.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum. Immunol. 2005, 66, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Panduro, M.; Benoist, C.; Mathis, D. Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2585–E2593. [Google Scholar] [CrossRef]
- Castiglioni, A.; Corna, G.; Rigamonti, E.; Basso, V.; Vezzoli, M.; Monno, A.; Almada, A.E.; Mondino, A.; Wagers, A.J.; Manfredi, A.A.; et al. FOXP3+ T Cells Recruited to Sites of Sterile Skeletal Muscle Injury Regulate the Fate of Satellite Cells and Guide Effective Tissue Regeneration. PLoS ONE 2015, 10, e0128094. [Google Scholar] [CrossRef] [PubMed]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 2016, 213, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Vetrone, S.A.; Montecino-Rodriguez, E.; Kudryashova, E.; Kramerova, I.; Hoffman, E.P.; Liu, S.D.; Miceli, M.C.; Spencer, M.J. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β. J. Clin. Investig. 2009, 119, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.M.; Blair, S.J.; Warunek, J.; Heffner, R.R.; Blader, I.J.; Wohlfert, E.A. Regulatory T Cells Promote Myositis and Muscle Damage in Toxoplasma gondii Infection. J. Immunol. 2017, 198, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Cosgrove, B.D.; Ho, A.T.V. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015, 21, 854. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Muñoz-Cánoves, P. The ins and outs of muscle stem cell aging. Skelet Muscle 2016, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Lages, C.S.; Suffia, I.; Velilla, P.A.; Huang, B.; Warshaw, G.; Hildeman, D.A.; Belkaid, Y.; Chougnet, C. Functional Regulatory T Cells Accumulate in Aged Hosts and Promote Chronic Infectious Disease Reactivation. J. Immunol. 2008, 181, 1835–1848. [Google Scholar] [CrossRef]
- Qazi, T.H.; Duda, G.N.; Ort, M.J.; Perka, C.; Geissler, S.; Winkler, T. Cell therapy to improve regeneration of skeletal muscle injuries. J. Cachexia Sarcopenia Muscle 2019, 10, 501–516. [Google Scholar] [CrossRef]
- Klimczak, A.; Kozlowska, U.; Kurpisz, M. Muscle Stem/Progenitor Cells and Mesenchymal Stem Cells of Bone Marrow for Skeletal Muscle Regeneration in Muscular Dystrophies. Arch. Immunol. Ther. Exp. 2018, 66, 13. [Google Scholar]
- Caseiro, A.; Pereira, T.; Bártolo, P.; Santos, J.; Luís, A.; Maurício, A.L.A.A.C. Trends in Mesenchymal Stem Cells’ Applications for Skeletal Muscle Repair and Regeneration; Demirer, T., Ed.; IntechOpen: London, UK, 2015. [Google Scholar]
- Winkler, T.; Perka, C.; von Roth, P.; Agres, A.N.; Plage, H.; Preininger, B.; Pumberger, M.; Geissler, S.; Hagai, E.L.; Ofir, R.; et al. Immunomodulatory placental-expanded, mesenchymal stromal cells improve muscle function following hip arthroplasty. J. Cachexia Sarcopenia Muscle 2018, 9, 880–897. [Google Scholar] [CrossRef]
- Hof-Nahor, I.; Leshansky, L.; Shivtiel, S.; Eldor, L.; Aberdam, D.; Itskovitz-Eldor, J.; Berrih-Aknin, S. Human mesenchymal stem cells shift CD8+ T cells towards a suppressive phenotype by inducing tolerogenic monocytes. J. Cell Sci. 2012, 125, 4640–4650. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef]
- Groh, M.E.; Maitra, B.; Szekely, E.; Koc, O.N. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp. Hematol. 2005, 33, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [PubMed]
- Helal, M.A.M.; Shaheen, N.E.M.; Abu Zahra, F.A. Immunomodulatory capacity of the local mesenchymal stem cells transplantation after severe skeletal muscle injury in female rats. Immunopharmacol. Immunotoxicol. 2016, 38, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, S.; Zhang, H.; Zhu, B.; Su, Y.; Zheng, C.; Tian, R.; Wang, M.; Kuang, H.; Zhao, X.; et al. Mesenchymal stem cells and extracellular matrix scaffold promote muscle regeneration by synergistically regulating macrophage polarization toward the M2 phenotype. Stem. Cell Res. Ther. 2018, 9, 88. [Google Scholar] [CrossRef]
- Corona, B.T.; Wu, X.; Ward, C.L.; McDaniel, J.S.; Rathbone, C.R.; Walters, T.J. The promotion of a functional fibrosis in skeletal muscle with volumetric muscle loss injury following the transplantation of muscle-ECM. Biomaterials 2013, 34, 3324–3335. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, A.; Zisa, D.; Leiker, M.; Johnston, C.; Lin, H.; Lee, T. Muscular dystrophy therapy by nonautologous mesenchymal stem cells: Muscle regeneration without immunosuppression and inflammation. Transplantation 2009, 87, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.H.; Dunn, A.J.; Talovic, M.; Haas, G.J.; Marcinczyk, M.; Elmashhady, H.; Kalaf, E.G.; Sell, S.A.; Garg, K. Aligned nanofibers of decellularized muscle ECM support myogenic activity in primary satellite cells in vitro. Biomed. Mater. 2019, 14, 035010. [Google Scholar] [CrossRef]
- Li, Y.; Jin, D.; Xie, W.; Wen, L.; Chen, W.; Xu, J.; Ding, J.; Ren, D.; Xiao, Z. Mesenchymal Stem Cells-Derived Exosomes: A Possible Therapeutic Strategy for Osteoporosis. Curr. Stem. Cell Res. Ther. 2018, 13, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Rezvani, K.; Shpall, E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transpl. 2019, 54, 789–792. [Google Scholar] [CrossRef]
- Qiu, G.; Zheng, G.; Ge, M.; Wang, J.; Huang, R.; Shu, Q.; Xu, J. Mesenchymal stem cell-derived extracellular vesicles affect disease outcomes via transfer of microRNAs. Stem. Cell Res. Ther. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, S.; Fierabracci, A. Insights into the Secretome of Mesenchymal Stem Cells and Its Potential Applications. Int. J. Mol. Sci. 2019, 20, 4597. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.W.; Wang, J.; Lee, C.J.; Liu, M.; Neelamegham, S.; Canty, J.M.; Nguyen, J. The microRNA regulatory landscape of MSC-derived exosomes: A systems view. Sci. Rep. 2018, 8, 1419. [Google Scholar] [CrossRef]
- Burrello, J.; Monticone, S.; Gai, C.; Gomez, Y.; Kholia, S.; Camussi, G. Stem Cell-Derived Extracellular Vesicles and Immune-Modulation. Front. Cell Dev. Biol. 2016, 4, 83. [Google Scholar] [CrossRef]
- Riau, A.K.; Ong, H.S.; Yam, G.H.F.; Mehta, J.S. Sustained Delivery System for Stem Cell-Derived Exosomes. Front. Pharm. 2019, 10, 1368. [Google Scholar] [CrossRef]
- Nakamura, Y.; Miyaki, S.; Ishitobi, H.; Matsuyama, S.; Nakasa, T.; Kamei, N.; Akimoto, T.; Higashi, Y.; Ochi, M. Mesenchymal-stem-cell-derived exosomes accelerate skeletal muscle regeneration. FEBS Lett. 2015, 589, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem. Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Magarotto, F.; Sgrò, A.; Dorigo Hochuli, A.H.; Andreetta, M.; Grassi, M.; Saggioro, M.; Nogara, L.; Tolomeo, A.M.; Francescato, R.; Collino, F.; et al. Muscle functional recovery is driven by extracellular vesicles combined with muscle extracellular matrix in a volumetric muscle loss murine model. Biomaterials 2021, 269, 120653. [Google Scholar] [CrossRef]
- Baer, A.; Colon-Moran, W.; Bhattarai, N. Characterization of the effects of immunomodulatory drug fingolimod (FTY720) on human T cell receptor signaling pathways. Sci. Rep. 2018, 8, 10910. [Google Scholar] [CrossRef]
- Thomson, A.W.; Bonham, C.A.; Zeevi, A. Mode of action of tacrolimus (FK506): Molecular and cellular mechanisms. Ther. Drug. Monit. 1995, 17, 584–591. [Google Scholar] [CrossRef]
- Heydemann, A. Severe murine limb-girdle muscular dystrophy type 2C pathology is diminished by FTY720 treatment. Muscle Nerve 2017, 56, 486–494. [Google Scholar] [CrossRef]
- Foster, A.D.; Vicente, D.; Sexton, J.J.; Johnston, L.; Clark, N.; Leonhardt, C.; Elster, E.A.; Davis, T.A.; Bradley, M.J. Administration of FTY720 during Tourniquet-Induced Limb Ischemia Reperfusion Injury Attenuates Systemic Inflammation. Mediat. Inflamm. 2017, 2017, 4594035. [Google Scholar] [CrossRef]
- Corona, B.T.; Rivera, J.C.; Wenke, J.C.; Greising, S.M. Tacrolimus as an adjunct to autologous minced muscle grafts for the repair of a volumetric muscle loss injury. J. Exp. Orthop. 2017, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orso, S.; Juan, A.H.; Ko, K.D.; Naz, F.; Perovanovic, J.; Gutierrez-Cruz, G.; Feng, X.; Sartorelli, V. Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development 2019, 146. [Google Scholar] [CrossRef]
- De Micheli, A.J.; Laurilliard, E.J.; Heinke, C.L.; Ravichandran, H.; Fraczek, P.; Soueid-Baumgarten, S.; De Vlaminck, I.; Elemento, O.; Cosgrove, B.D. Single-Cell Analysis of the Muscle Stem Cell Hierarchy Identifies Heterotypic Communication Signals Involved in Skeletal Muscle Regeneration. Cell Rep. 2020, 30, 3583–3595.e3585. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, A.B.; Smith, G.R.; Raue, U.; Begue, G.; Minchev, K.; Ruf-Zamojski, F.; Nair, V.D.; Wang, X.; Zhou, L.; Zaslavsky, E.; et al. Single-cell transcriptional profiles in human skeletal muscle. Sci. Rep. 2020, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- De Micheli, A.J.; Spector, J.A.; Elemento, O.; Cosgrove, B.D. A reference single-cell transcriptomic atlas of human skeletal muscle tissue reveals bifurcated muscle stem cell populations. Skelet. Muscle 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziemkiewicz, N.; Hilliard, G.; Pullen, N.A.; Garg, K. The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2021, 22, 3265. https://doi.org/10.3390/ijms22063265
Ziemkiewicz N, Hilliard G, Pullen NA, Garg K. The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. International Journal of Molecular Sciences. 2021; 22(6):3265. https://doi.org/10.3390/ijms22063265
Chicago/Turabian StyleZiemkiewicz, Natalia, Genevieve Hilliard, Nicholas A. Pullen, and Koyal Garg. 2021. "The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration" International Journal of Molecular Sciences 22, no. 6: 3265. https://doi.org/10.3390/ijms22063265
APA StyleZiemkiewicz, N., Hilliard, G., Pullen, N. A., & Garg, K. (2021). The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. International Journal of Molecular Sciences, 22(6), 3265. https://doi.org/10.3390/ijms22063265