
Genetics of Azoospermia


Abstract
1. Introduction
2. Chromosomal Anomalies Causing Azoospermia
2.1. Karyotype Anomalies
2.1.1. Klinefelter Syndrome (47,XXY)
2.1.2. 46,XX Testicular/Ovo-Testicular Disorder of Sex Development
2.1.3. Yq–
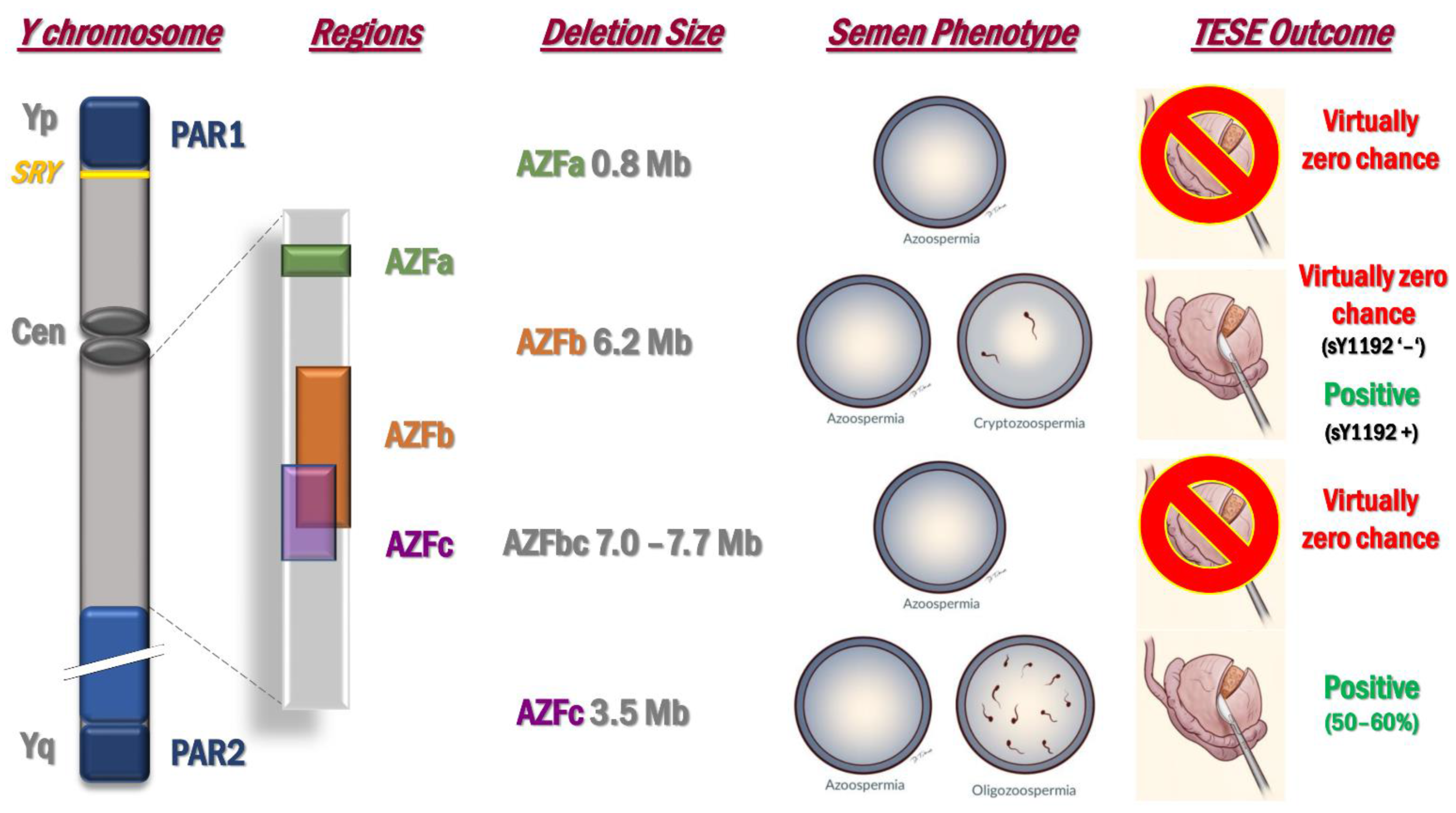
2.2. Microdeletions of the Y Chromosome: AZF Deletions
3. Monogenic Forms of Azoospermia
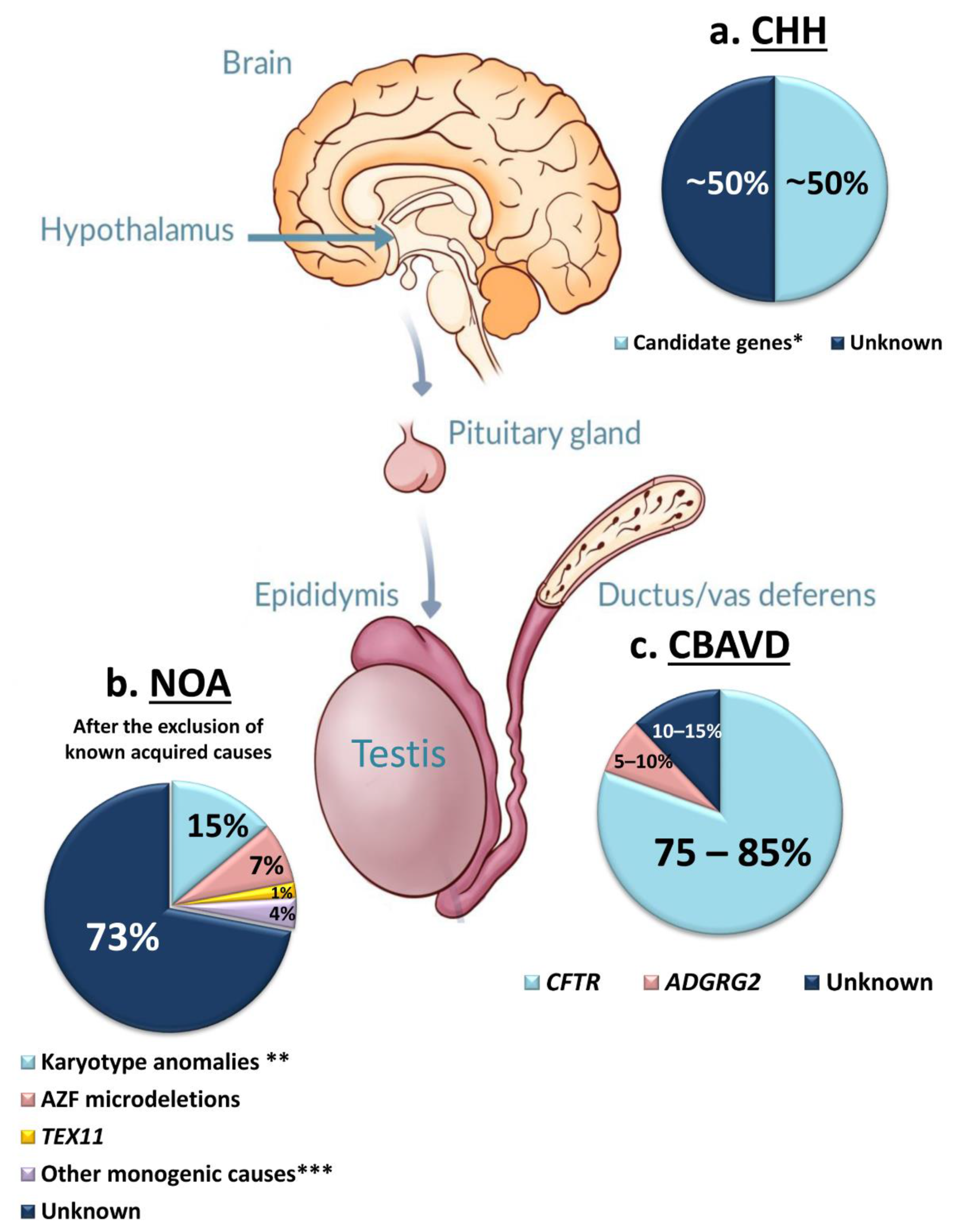
3.1. Congenital Hypogonadotropic Hypogonadism (CHH)
3.2. Congenital Bilateral Absence of Vas Deferens (CBAVD)
3.3. Primary Testicular Failure
3.3.1. Candidate Genes for the SCOS Phenotype
3.3.2. Candidate Genes for the MA Phenotype
3.3.3. Candidate Genes Associated with Different Types of Testicular Phenotype
3.3.4. Candidate Genes for iNOA with Undefined Testicular Phenotype
4. Common Monogenic Defects in Male and Female Primary Gonadal Failure
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forti, G.; Krausz, C. Clinical review 100: Evaluation and treatment of the infertile couple. J. Clin. Endocrinol. Metab. 1998, 83, 4177–4188. [Google Scholar]
- Lotti, F.; Maggi, M. Sexual dysfunction and male infertility. Nat. Rev. Urol. 2018, 15, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Tournaye, H.; Krausz, C.; Oates, R.D. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. 2017, 5, 544–553. [Google Scholar] [CrossRef]
- Krausz, C.; Cioppi, F.; Riera-Escamilla, A. Testing for genetic contributions to infertility: Potential clinical impact. Expert Rev. Mol. Diagn. 2018, 18, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Riera-Escamilla, A. Genetics of male infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Hoefsloot, L.; Simoni, M.; Tüttelmann, F.; European Academy of Andrology. European Molecular Genetics Quality Network EAA/EMQN best practice guidelines for molecular diagnosis of Y-chromosomal microdeletions: State-of-the-art 2013. Andrology 2014, 2, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, B.; Swee, D.S.; Quinton, R.; Bonomi, M. Genetics of congenital hypogonadotropic hypogonadism: Peculiarities and phenotype of an oligogenic disease. Hum. Genet. 2021, 140, 77–111. [Google Scholar] [CrossRef]
- Butz, H.; Nyírő, G.; Kurucz, P.A.; Likó, I.; Patócs, A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: From panel design towards result interpretation in clinical practice. Hum. Genet. 2021, 140, 113–134. [Google Scholar] [CrossRef]
- Oud, M.S.; Volozonoka, L.; Smits, R.M.; Vissers, L.E.L.M.; Ramos, L.; Veltman, J.A. A systematic review and standardized clinical validity assessment of male infertility genes. Hum. Reprod. 2019, 34, 932–941. [Google Scholar] [CrossRef]
- Krausz, C.; Riera-Escamilla, A.; Moreno-Mendoza, D.; Holleman, K.; Cioppi, F.; Algaba, F.; Pybus, M.; Friedrich, C.; Wyrwoll, M.J.; Casamonti, E.; et al. Genetic dissection of spermatogenic arrest through exome analysis: Clinical implications for the management of azoospermic men. Genet. Med. 2020, 22, 1956–1966. [Google Scholar] [CrossRef]
- Kasak, L.; Laan, M. Monogenic causes of non-obstructive azoospermia: Challenges, established knowledge, limitations and perspectives. Hum. Genet. 2021, 140, 135–154. [Google Scholar] [CrossRef]
- Klinefelter, H.F.; Reifenstein, E.C.; Albright, F. Syndrome characterized by gynecomastia, aspermatogenesis without A-Leydigism, and increased excretion of follicle-stimulating hormone1. J. Clin. Endocrinol. Metab. 1942, 2, 615–627. [Google Scholar] [CrossRef]
- Zitzmann, M.; Aksglaede, L.; Corona, G.; Isidori, A.M.; Juul, A.; T’Sjoen, G.; Kliesch, S.; D’Hauwers, K.; Toppari, J.; Słowikowska-Hilczer, J.; et al. European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology. Andrology 2021, 9, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Vloeberghs, V.; Verheyen, G.; Santos-Ribeiro, S.; Staessen, C.; Verpoest, W.; Gies, I.; Tournaye, H. Is genetic fatherhood within reach for all azoospermic Klinefelter men? PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Abramsky, L.; Chapple, J. 47,XXY (Klinefelter Syndrome) and 47,XYY: Estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat. Diagn. 1997, 17, 363–368. [Google Scholar] [CrossRef]
- Bojesen, A.; Juul, S.; Gravholt, C.H. Prenatal and postnatal prevalence of Klinefelter syndrome: A national registry study. J. Clin. Endocrinol. Metab. 2003, 88, 622–626. [Google Scholar] [CrossRef]
- Bojesen, A.; Gravholt, C.H. Klinefelter syndrome in clinical practice. Nat. Clin. Pract. Urol. 2007, 4, 192–204. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Chang, S.; Wallentin, M.; Fedder, J.; Moore, P.; Skakkebæk, A. Klinefelter syndrome: Integrating genetics, neuropsychology, and endocrinology. Endocr. Rev. 2018, 39, 389–423. [Google Scholar] [CrossRef]
- Ottesen, A.M.; Aksglaede, L.; Garn, I.; Tartaglia, N.; Tassone, F.; Gravholt, C.H.; Bojesen, A.; Sørensen, K.; Jørgensen, N.; De Meyts, E.R.; et al. Increased number of sex chromosomes affects height in a nonlinear fashion: A study of 305 patients with sex chromosome aneuploidy. Am. J. Med. Genet. Part A 2010, 152, 1206–1212. [Google Scholar] [CrossRef]
- Zitzmann, M.; Depenbusch, M.; Jörg Gromoll, A.; Nieschlag, E. X-chromosome inactivation patterns and androgen receptor functionality influence phenotype and social characteristics as well as pharmacogenetics of testosterone therapy in klinefelter patients. J. Clin. Endocrinol. Metab. 2004, 89, 6208–6217. [Google Scholar] [CrossRef]
- Bojesen, A.; Hertz, J.M.; Gravholt, C.H. Genotype and phenotype in Klinefelter syndrome-impact of androgen receptor polymorphism and skewed X inactivation. Int. J. Androl. 2011, 34. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Skakkebæk, A.; Trolle, C.; Bojesen, A.; Hertz, J.M.; Cohen, A.; Hougaard, D.M.; Wallentin, M.; Pedersen, A.D.; Østergaard, J.R.; et al. Anthropometry in klinefelter syndrome-multifactorial influences due to CAG length, testosterone treatment and possibly intrauterine hypogonadism. J. Clin. Endocrinol. Metab. 2015, 100, E508–E517. [Google Scholar] [CrossRef]
- Belling, K.; Russo, F.; Jensen, A.B.; Dalgaard, M.D.; Westergaard, D.; De Meyts, E.R.; Skakkebaek, N.E.; Juul, A.; Brunak, S. Klinefelter syndrome comorbidities linked to increased X chromosome gene dosage and altered protein interactome activity. Hum. Mol. Genet. 2017, 26, 1219–1229. [Google Scholar] [CrossRef]
- Sharma, A.; Jamil, M.A.; Nuesgen, N.; Schreiner, F.; Priebe, L.; Hoffmann, P.; Herns, S.; Nöthen, M.M.; Fröhlich, H.; Oldenburg, J.; et al. DNA methylation signature in peripheral blood reveals distinct characteristics of human X chromosome numerical aberrations. Clin. Epigenetics 2015, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Viana, J.; Pidsley, R.; Troakes, C.; Spiers, H.; Wong, C.C.Y.; Al-Sarraj, S.; Craig, I.; Schalkwyk, L.; Mill, J. Epigenomic and transcriptomic signatures of a Klinefelter syndrome (47,XXY) karyotype in the brain. Epigenetics 2014, 9, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Stochholm, K.; Juul, S.; Gravholt, C.H. Poor socio-economic status in 47,XXX-An unexpected effect of an extra X chromosome. Eur. J. Med. Genet. 2013, 56, 286–291. [Google Scholar] [CrossRef]
- Davis, S.M.; Rogol, A.D.; Ross, J.L. Testis Development and fertility potential in boys with Klinefelter syndrome. Endocrinol. Metab. Clin. N. Am. 2015, 44, 843–865. [Google Scholar] [CrossRef] [PubMed]
- Levron, J.; Aviram-Goldring, A.; Madgar, I.; Raviv, G.; Barkai, G.; Dor, J. Sperm chromosome analysis and outcome of IVF in patients with non-mosaic Klinefelter’s syndrome. Fertil. Steril. 2000, 74, 925–929. [Google Scholar] [CrossRef]
- Sciurano, R.B.; Luna Hisano, C.V.; Rahn, M.I.; Brugo Olmedo, S.; Rey Valzacchi, G.; Coco, R.; Solari, A.J. Focal spermatogenesis originates in euploid germ cells in classical Klinefelter patients. Hum. Reprod. 2009, 24, 2353–2360. [Google Scholar] [CrossRef]
- Corona, G.; Pizzocaro, A.; Lanfranco, F.; Garolla, A.; Pelliccione, F.; Vignozzi, L.; Ferlin, A.; Foresta, C.; Jannini, E.A.; Maggi, M.; et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2017, 23, 1–11. [Google Scholar] [CrossRef]
- Rohayem, J.; Fricke, R.; Czeloth, K.; Mallidis, C.; Wistuba, J.; Krallmann, C.; Zitzmann, M.; Kliesch, S. Age and markers of Leydig cell function, but not of Sertoli cell function predict the success of sperm retrieval in adolescents and adults with Klinefelter’s syndrome. Andrology 2015, 3, 868–875. [Google Scholar] [CrossRef]
- Gies, I.; Tournaye, H.; De Schepper, J. Attitudes of parents of Klinefelter boys and pediatricians towards neonatal screening and fertility preservation techniques in Klinefelter syndrome. Eur. J. Pediatr. 2016, 175, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Scarselli, F.; Minasi, M.G.; Casciani, V.; Zavaglia, D.; Dente, D.; Tesarik, J.; Franco, G. Birth of 16 healthy children after ICSI in cases of nonmosaic Klinefelter syndrome. Hum. Reprod. 2013, 28, 1155–1160. [Google Scholar] [CrossRef]
- Fullerton, G.; Hamilton, M.; Maheshwari, A. Should non-mosaic Klinefelter syndrome men be labelled as infertile in 2009? Hum. Reprod. 2010, 25, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Denschlag, D.; Tempfer, C.; Kunze, M.; Wolff, G.; Keck, C. Assisted reproductive techniques in patients with Klinefelter syndrome: A critical review. Fertil. Steril. 2004, 82, 775–779. [Google Scholar] [CrossRef]
- Maiburg, M.; Repping, S.; Giltay, J. The genetic origin of Klinefelter syndrome and its effect on spermatogenesis. Fertil. Steril. 2012, 98, 253–260. [Google Scholar] [CrossRef] [PubMed]
- De la Chapelle, A.; Hortling, H.; Niemi, M.; Wennström, J. XX Sex Chromosomes in a Human Male. Acta Med. Scand. 1964, 175, 25–38. [Google Scholar] [CrossRef]
- Vorona, E.; Zitzmann, M.; Gromoll, J.; Schüring, A.N.; Nieschlag, E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J. Clin. Endocrinol. Metab. 2007, 92, 3458–3465. [Google Scholar] [CrossRef]
- Kousta, E.; Papathanasiou, A.; Skordis, N. Sex determination and disorders of sex development according to the revised nomenclature and classification in 46,XX individuals. Hormones 2010, 9, 218–231. [Google Scholar] [CrossRef]
- McElreavey, K.; Vilain, E.; Abbas, N.; Herskowitz, I.; Fellous, M. A regulatory cascade hypothesis for mammalian sex determination: SRY represses a negative regulator of male development. Proc. Natl. Acad. Sci. USA 1993, 90, 3368–3372. [Google Scholar] [CrossRef]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.-S.; Wu, Y.-N.; Wu, C.-C.; Hwang, J.-L. Cytogenic and molecular analyses of 46,XX male syndrome with clinical comparison to other groups with testicular azoospermia of genetic origin. J. Formos. Med. Assoc. 2013, 112, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.M.; Vilain, E. Translational genetics for diagnosis of human disorders of sex development. Annu. Rev. Genomics Hum. Genet. 2013, 14, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Skaletsky, H.; Kuroda-Kawaguchi, T.; Minx, P.J.; Cordum, H.S.; Hillier, L.; Brown, L.G.; Repping, S.; Pyntikova, T.; Ali, J.; Bieri, T.; et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003, 423, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Costa, P. Sex, rebellion and decadence: The scandalous evolutionary history of the human Y chromosome. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1851–1863. [Google Scholar] [CrossRef][Green Version]
- Tiepolo, L.; Zuffardi, O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum. Genet. 1976, 34, 119–124. [Google Scholar] [CrossRef]
- Vogt, P.H.; Edelmann, A.; Kirsch, S.; Henegariu, O.; Hirschmann, P.; Kiesewetter, F.; Köhn, F.M.; Schill, W.B.; Farah, S.; Ramos, C.; et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum. Mol. Genet. 1996, 5, 933–943. [Google Scholar] [CrossRef]
- Turner, J.M.A.; Mahadevaiah, S.K.; Fernandez-Capetillo, O.; Nussenzweig, A.; Xu, X.; Deng, C.X.; Burgoyne, P.S. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat. Genet. 2005, 37, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Casamonti, E. Spermatogenic failure and the Y chromosome. Hum. Genet. 2017, 136, 637–655. [Google Scholar] [CrossRef]
- Lo Giacco, D.; Chianese, C.; Sánchez-Curbelo, J.; Bassas, L.; Ruiz, P.; Rajmil, O.; Sarquella, J.; Vives, A.; Ruiz-Castañé, E.; Oliva, R.; et al. Clinical relevance of Y-linked CNV screening in male infertility: New insights based on the 8-year experience of a diagnostic genetic laboratory. Eur. J. Hum. Genet. 2014, 22, 754–761. [Google Scholar] [CrossRef]
- Ginalski, K.; Rychlewski, L.; Baker, D.; Grishin, N.V. Protein structure prediction for the male-specific region of the human Y chromosome. Proc. Natl. Acad. Sci. USA 2004, 101, 2305–2310. [Google Scholar] [CrossRef] [PubMed]
- Tyler-Smith, C.; Krausz, C. The will-o’-the-wisp of genetics—Hunting for the azoospermia factor gene. N. Engl. J. Med. 2009, 360, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Stryker, J.M.; Lambowitz, A.M. A DEAD-Box protein functions as an ATP-dependent RNA chaperone in group I intron splicing. Cell 2002, 109, 769–779. [Google Scholar] [CrossRef]
- Wu, Q.; Chen, G.W.; Yan, T.F.; Wang, H.; Liu, Y.L.; Li, Z.; Duan, S.W.; Sun, F.; Feng, Y.; Shi, H.J. Prevalent false positives of azoospermia factor a (AZFa) microdeletions caused by single-nucleotide polymorphism rs72609647 in the sY84 screening of male infertility. Asian J. Androl. 2011, 13, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Stouffs, K.; Vloeberghs, V.; Gheldof, A.; Tournaye, H.; Seneca, S. Are AZFb deletions always incompatible with sperm production? Andrology 2017, 5, 691–694. [Google Scholar] [CrossRef]
- Flannigan, R.; Schlegel, P.N. Genetic diagnostics of male infertility in clinical practice. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 44, 26–37. [Google Scholar] [CrossRef]
- Jaruzelska, J.; Korcz, A.; Wojda, A.; Jedrzejczak, P.; Bierla, J.; Surmacz, T.; Pawelczyk, L.; Page, D.C.; Kotecki, M. Mosaicism for 45,X cell line may accentuate the severity of spermatogenic defects in men with AZFc deletion. J. Med. Genet. 2001, 38, 798–802. [Google Scholar] [CrossRef]
- Siffroi, J.P.; Le Bourhis, C.; Krausz, C.; Barbaux, S.; Quintana-Murci, L.; Kanafani, S.; Rouba, H.; Bujan, L.; Bourrouillou, G.; Seifer, I.; et al. Sex chromosome mosaicism in males carrying Y chromosome long arm deletions. Hum. Reprod. 2000, 15, 2559–2562. [Google Scholar] [CrossRef]
- Patsalis, P.C.; Skordis, N.; Sismani, C.; Kousoulidou, L.; Koumbaris, G.; Eftychi, C.; Stavrides, G.; Ioulianos, A.; Kitsiou-Tzeli, S.; Galla-Voumvouraki, A.; et al. Identification of high frequency of Y chromosome deletions in patients with sex chromosome mosaicism and correlation with the clinical phenotype and Y-chromosome instability. Am. J. Med. Genet. 2005, 135, 145–149. [Google Scholar] [CrossRef]
- Krausz, C.; Degl’Innocenti, S. Y chromosome and male infertility: Update, 2006. Front. Biosci. 2006, 11, 3049–3061. [Google Scholar] [CrossRef]
- Le Bourhis, C.; Siffroi, J.P.; McElreavey, K.; Dadoune, J.P. Y chromosome microdeletions and germinal mosaicism in infertile males. Mol. Hum. Reprod. 2000, 6, 688–693. [Google Scholar] [CrossRef]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamäki, J.; Raivio, T.; Pitteloud, N. Clinical management of congenital hypogonadotropic hypogonadism. Endocr. Rev. 2019, 40, 669–710. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Bouloux, P.-M.; Dattani, M.T.; de Roux, N.; Dodé, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.-P.; Juul, A.; et al. European Consensus Statement on congenital hypogonadotropic hypogonadism—Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, A.A.; Raivio, T.; Pitteloud, N. Reversible hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2016, 174, R267–R274. [Google Scholar] [CrossRef] [PubMed]
- Bieth, E.; Hamdi, S.M.; Mieusset, R. Genetics of the congenital absence of the vas deferens. Hum. Genet. 2021, 140, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Oates, R.D.; Amos, J.A. The genetic basis of congenital bilateral absence of the vas deferens and cystic fibrosis. J. Androl. 1994, 15, 1–8. [Google Scholar]
- Bergougnoux, A.; Délétang, K.; Pommier, A.; Varilh, J.; Houriez, F.; Altieri, J.P.; Koenig, M.; Férec, C.; Claustres, M.; Lalau, G.; et al. Functional characterization and phenotypic spectrum of three recurrent disease-causing deep intronic variants of the CFTR gene. J. Cyst. Fibros. 2019, 18, 468–475. [Google Scholar] [CrossRef]
- Feng, J.; Wu, X.; Zhang, Y.; Yang, X.; Ma, G.; Chen, S.; Luo, S.; Zhang, Y. A novel mutation (−195C>A) in the promoter region of CFTR gene is associated with Chinese Congenital Bilateral Absence of Vas Deferens (CBAVD). Gene 2019, 719. [Google Scholar] [CrossRef]
- De Souza, D.A.S.; Faucz, F.R.; Pereira-Ferrari, L.; Sotomaior, V.S.; Raskin, S. Congenital bilateral absence of the vas deferens as an atypical form of cystic fibrosis: Reproductive implications and genetic counseling. Andrology 2018, 6, 127–135. [Google Scholar] [CrossRef]
- Deignan, J.L.; Astbury, C.; Cutting, G.R.; del Gaudio, D.; Gregg, A.R.; Grody, W.W.; Monaghan, K.G.; Richards, S. CFTR variant testing: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1288–1295. [Google Scholar] [CrossRef]
- Obermann, H.; Samalecos, A.; Osterhoff, C.; Schröder, B.; Heller, R.; Kirchhoff, C. HE6, a two-subunit heptahelical receptor associated with apical membranes of efferent and epididymal duct epithelia. Mol. Reprod. Dev. 2003, 64, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Patat, O.; Pagin, A.; Siegfried, A.; Mitchell, V.; Chassaing, N.; Faguer, S.; Monteil, L.; Gaston, V.; Bujan, L.; Courtade-Saïdi, M.; et al. Truncating mutations in the adhesion g protein-coupled receptor G2 Gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am. J. Hum. Genet. 2016, 99, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Baumann, C.; Kirchhoff, C.; Ivell, R.; Nubbemeyer, R.; Habenicht, U.-F.; Theuring, F.; Gottwald, U. targeted deletion of the epididymal receptor HE6 results in fluid dysregulation and male infertility. Mol. Cell. Biol. 2004, 24, 8642–8648. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.; Zhang, W.; Pan, H.; Li, T.; Liu, B.; Li, H.; Wang, B. Pathogenic role of ADGRG2 in CBAVD patients replicated in Chinese population. Andrology 2017, 5, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.J.; Pollock, N.; Jiang, H.; Castro, C.; Nazli, R.; Ahmed, J.; Basit, S.; Rajkovic, A.; Yatsenko, A.N. X-linked ADGRG2 mutation and obstructive azoospermia in a large Pakistani family. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Yuan, P.; Liang, Z.K.; Liang, H.; Zheng, L.Y.; Li, D.; Li, J.; Zhang, J.; Tian, J.; Lai, L.H.; Zhang, K.; et al. Expanding the phenotypic and genetic spectrum of Chinese patients with congenital absence of vas deferens bearing CFTR and ADGRG2 alleles. Andrology 2019, 7, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Pagin, A.; Bergougnoux, A.; Girodon, E.; Reboul, M.P.; Willoquaux, C.; Kesteloot, M.; Raynal, C.; Bienvenu, T.; Humbert, M.; Lalau, G.; et al. Novel ADGRG2 truncating variants in patients with X-linked congenital absence of vas deferens. Andrology 2020, 8, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-N.; Lin, Y.-H.; Chiang, H.-S. SLC9A3 is a novel pathogenic gene in Taiwanese males with congenital bilateral absence of the vas deferens. Eur. Urol. Suppl. 2018, 17, e1092. [Google Scholar] [CrossRef]
- Wu, Y.N.; Chen, K.C.; Wu, C.C.; Lin, Y.H.; Chiang, H.S. SLC9A3 affects vas deferens development and associates with taiwanese congenital bilateral absence of the vas deferens. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Chalmel, F.; Lardenois, A.; Evrard, B.; Mathieu, R.; Feig, C.; Demougin, P.; Gattiker, A.; Schulze, W.; Jégou, B.; Kirchhoff, C.; et al. Global human tissue profiling and protein network analysis reveals distinct levels of transcriptional germline-specificity and identifies target genes for male infertility. Hum. Reprod. 2012, 27, 3233–3248. [Google Scholar] [CrossRef]
- Soraggi, S.; Riera, M.; Rajpert-De Meyts, E.; Schierup, M.H.; Almstrup, K. Evaluating genetic causes of azoospermia: What can we learn from a complex cellular structure and single-cell transcriptomics of the human testis? Hum. Genet. 2021, 140, 183–201. [Google Scholar] [CrossRef]
- Krausz, C.; Riera-Escamilla, A.; Chianese, C.; Moreno-Mendoza, D.; Ars, E.; Rajmil, O.; Pujol, R.; Bogliolo, M.; Blanco, I.; Rodríguez, I.; et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet. Med. 2019, 21, 189–194. [Google Scholar] [CrossRef]
- Miyamoto, T.; Bando, Y.; Koh, E.; Tsujimura, A.; Miyagawa, Y.; Iijima, M.; Namiki, M.; Shiina, M.; Ogata, K.; Matsumoto, N.; et al. A PLK4 mutation causing azoospermia in a man with Sertoli cell-only syndrome. Andrology 2016, 4, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fakhro, K.A.; Elbardisi, H.; Arafa, M.; Robay, A.; Rodriguez-Flores, J.L.; Al-Shakaki, A.; Syed, N.; Mezey, J.G.; Abi Khalil, C.; Malek, J.A.; et al. Point-of-care whole-exome sequencing of idiopathic male infertility. Genet. Med. 2018, 20, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Jiao, Y.; Khan, R.; Jiang, X.; Javed, A.R.; Ali, A.; Zhang, H.; Zhou, J.; Naeem, M.; Murtaza, G.; et al. Homozygous mutations in C14orf39/SIX6OS1 cause non-obstructive azoospermia and premature ovarian insufficiency in humans. Am. J. Hum. Genet. 2021, 108. [Google Scholar] [CrossRef]
- He, W.B.; Tu, C.F.; Liu, Q.; Meng, L.L.; Yuan, S.M.; Luo, A.X.; He, F.S.; Shen, J.; Li, W.; Du, J.; et al. DMC1 mutation that causes human non-obstructive azoospermia and premature ovarian insufficiency identified by whole-exome sequencing. J. Med. Genet. 2018, 55, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Salas-Huetos, A.; Tüttelmann, F.; Wyrwoll, M.J.; Kliesch, S.; Lopes, A.M.; Conçalves, J.; Boyden, S.E.; Wöste, M.; Hotaling, J.M.; Aston, K.I.; et al. Disruption of human meiotic telomere complex genes TERB1, TERB2 and MAJIN in men with non-obstructive azoospermia. Hum. Genet. 2020, 140, 217–227. [Google Scholar] [CrossRef]
- Ben Khelifa, M.; Ghieh, F.; Boudjenah, R.; Hue, C.; Fauvert, D.; Dard, R.; Garchon, H.J.; Vialard, F. A MEI1 homozygous missense mutation associated with meiotic arrest in a consanguineous family. Hum. Reprod. 2018, 33, 1034–1037. [Google Scholar] [CrossRef]
- Nguyen, N.M.P.; Ge, Z.J.; Reddy, R.; Fahiminiya, S.; Sauthier, P.; Bagga, R.; Sahin, F.I.; Mahadevan, S.; Osmond, M.; Breguet, M.; et al. Causative mutations and mechanism of androgenetic hydatidiform moles. Am. J. Hum. Genet. 2018, 103, 740–751. [Google Scholar] [CrossRef]
- Gershoni, M.; Hauser, R.; Yogev, L.; Lehavi, O.; Azem, F.; Yavetz, H.; Pietrokovski, S.; Kleiman, S.E. A familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genes. Genet. Med. 2017, 19, 998–1006. [Google Scholar] [CrossRef]
- Gershoni, M.; Hauser, R.; Barda, S.; Lehavi, O.; Arama, E.; Pietrokovski, S.; Kleiman, S.E. A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum. Reprod. 2019, 34, 666–671. [Google Scholar] [CrossRef]
- Riera-Escamilla, A.; Enguita-Marruedo, A.; Moreno-Mendoza, D.; Chianese, C.; Sleddens-Linkels, E.; Contini, E.; Benelli, M.; Natali, A.; Colpi, G.M.; Ruiz-Castañé, E.; et al. Sequencing of a “mouse azoospermia” gene panel in azoospermic men: Identification of RNF212 and STAG3 mutations as novel genetic causes of meiotic arrest. Hum. Reprod. 2019, 34, 978–988. [Google Scholar] [CrossRef]
- Becherel, O.J.; Fogel, B.L.; Zeitlin, S.I.; Samaratunga, H.; Greaney, J.; Homer, H.; Lavin, M.F. Disruption of spermatogenesis and infertility in ataxia with oculomotor apraxia type 2 (AOA2). Cerebellum 2019, 18, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Catford, S.R.; O’Bryan, M.K.; McLachlan, R.I.; Delatycki, M.B.; Rombauts, L. Germ cell arrest associated with aSETX mutation in ataxia oculomotor apraxia type 2. Reprod. Biomed. Online 2019, 38, 961–965. [Google Scholar] [CrossRef]
- Yao, C.; Yang, C.; Zhao, L.; Li, P.; Tian, R.; Chen, H.; Guo, Y.; Huang, Y.; Zhi, E.; Zhai, J.; et al. Bi-allelic SHOC1 loss-of-function mutations cause meiotic arrest and non-obstructive azoospermia. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kherraf, Z.-E.; Christou-Kent, M.; Karaouzene, T.; Amiri-Yekta, A.; Martinez, G.; Vargas, A.S.; Lambert, E.; Borel, C.; Dorphin, B.; Aknin-Seifer, I.; et al. SPINK2 deficiency causes infertility by inducing sperm defects in heterozygotes and azoospermia in homozygotes. EMBO Mol. Med. 2017, 9, 1132–1149. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, S.; McElreavy, K.; Robevska, G.; Akloul, L.; Ghieh, F.; Sreenivasan, R.; Beaumont, M.; Bashamboo, A.; Bignon-Topalovic, J.; Neyroud, A.S.; et al. STAG3 homozygous missense variant causes primary ovarian insufficiency and male non-obstructive azoospermia. Mol. Hum. Reprod. 2020, 26, 665–677. [Google Scholar] [CrossRef]
- Van Der Bijl, N.; Röpke, A.; Biswas, U.; Wöste, M.; Jessberger, R.; Kliesch, S.; Friedrich, C.; Tüttelmann, F. Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum. Reprod. 2019, 34, 2112–2119. [Google Scholar] [CrossRef]
- Nakamura, S.; Kobori, Y.; Ueda, Y.; Tanaka, Y.; Ishikawa, H.; Yoshida, A.; Katsumi, M.; Saito, K.; Nakamura, A.; Ogata, T.; et al. STX2 is a causative gene for nonobstructive azoospermia. Hum. Mutat. 2018, 39, 830–833. [Google Scholar] [CrossRef]
- Pashaei, M.; Rahimi Bidgoli, M.M.; Zare-Abdollahi, D.; Najmabadi, H.; Haji-Seyed-Javadi, R.; Fatehi, F.; Alavi, A. The second mutation of SYCE1 gene associated with autosomal recessive nonobstructive azoospermia. J. Assist. Reprod. Genet. 2020, 37, 451–458. [Google Scholar] [CrossRef]
- Maor-Sagie, E.; Cinnamon, Y.; Yaacov, B.; Shaag, A.; Goldsmidt, H.; Zenvirt, S.; Laufer, N.; Richler, C.; Frumkin, A. Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J. Assist. Reprod. Genet. 2015, 32, 887–891. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Tu, C.; Meng, L.; Yuan, S.; Sjaarda, C.; Luo, A.; Du, J.; Li, W.; Gong, F.; Zhong, C.; et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet. Med. 2019, 21, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Zheng, L.; Ji, Z.; Mei, L.; Ding, L.; Lin, S.; Wang, X.; Yang, X.; Li, P. A novel TEX11 mutation induces azoospermia: A case report of infertile brothers and literature review. BMC Med. Genet. 2018, 19, 1–7. [Google Scholar] [CrossRef]
- Yang, F.; Silber, S.; Leu, N.A.; Oates, R.D.; Marszalek, J.D.; Skaletsky, H.; Brown, L.G.; Rozen, S.; Page, D.C.; Wang, P.J. TEX11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol. Med. 2015, 7, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Yatsenko, A.N.; Georgiadis, A.P.; Röpke, A.; Berman, A.J.; Jaffe, T.; Olszewska, M.; Westernströer, B.; Sanfilippo, J.; Kurpisz, M.; Rajkovic, A.; et al. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N. Engl. J. Med. 2015, 372, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, J.; Dai, L.; Zhu, Y.; Hu, H.; Tan, L.; Chen, W.; Liang, D.; He, J.; Tu, M.; et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J. Med. Genet. 2018, 55, 628–636. [Google Scholar] [CrossRef]
- Ayhan, Ö.; Balkan, M.; Guven, A.; Hazan, R.; Atar, M.; Tok, A.; Tolun, A. Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J. Med. Genet. 2014, 51, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.M.; Aston, K.I.; Thompson, E.; Carvalho, F.; Gonçalves, J.; Huang, N.; Matthiesen, R.; Noordam, M.J.; Quintela, I.; Ramu, A.; et al. Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Yin, H.; Ma, H.; Hussain, S.; Zhang, H.; Xie, X.; Jiang, L.; Jiang, X.; Iqbal, F.; Bukhari, I.; Jiang, H.; et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet. Med. 2019, 21, 62–70. [Google Scholar] [CrossRef]
- Kasak, L.; Punab, M.; Nagirnaja, L.; Grigorova, M.; Minajeva, A.; Lopes, A.M.; Punab, A.M.; Aston, K.I.; Carvalho, F.; Laasik, E.; et al. Bi-allelic Recessive Loss-of-Function Variants in FANCM Cause Non-obstructive Azoospermia. Am. J. Hum. Genet. 2018, 103, 200–212. [Google Scholar] [CrossRef]
- Wyrwoll, M.J.; Temel, Ş.G.; Nagirnaja, L.; Oud, M.S.; Lopes, A.M.; van der Heijden, G.W.; Heald, J.S.; Rotte, N.; Wistuba, J.; Wöste, M.; et al. Bi-allelic mutations in M1AP are a frequent cause of meiotic arrest and severely impaired spermatogenesis leading to male infertility. Am. J. Hum. Genet. 2020, 107, 342–351. [Google Scholar] [CrossRef]
- Ferlin, A.; Rocca, M.S.; Vinanzi, C.; Ghezzi, M.; Di Nisio, A.; Foresta, C. Mutational screening of NR5A1 gene encoding steroidogenic factor 1 in cryptorchidism and male factor infertility and functional analysis of seven undescribed mutations. Fertil. Steril. 2015, 104, 163–169.e1. [Google Scholar] [CrossRef]
- Zare-Abdollahi, D.; Safari, S.; Mirfakhraie, R.; Movafagh, A.; Bastami, M.; Azimzadeh, P.; Salsabili, N.; Ebrahimizadeh, W.; Salami, S.; Omrani, M.D. Mutational screening of the NR5A1 in azoospermia. Andrologia 2015, 47, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Röpke, A.; Tewes, A.C.; Gromoll, J.; Kliesch, S.; Wieacker, P.; Tüttelmann, F. Comprehensive sequence analysis of the NR5A1 gene encoding steroidogenic factor 1 in a large group of infertile males. Eur. J. Hum. Genet. 2013, 21, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Bashamboo, A.; Ferraz-De-Souza, B.; Loureno, D.; Lin, L.; Sebire, N.J.; Montjean, D.; Bignon-Topalovic, J.; Mandelbaum, J.; Siffroi, J.P.; Christin-Maitre, S.; et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am. J. Hum. Genet. 2010, 87, 505–512. [Google Scholar] [CrossRef]
- Safari, S.; Zare-Abdollahi, D.; Mirfakhraie, R.; Ghafouri-Fard, S.; Pouresmaeili, F.; Movafagh, A.; Omrani, M.D. An Iranian family with azoospermia and premature ovarian insufficiency segregating NR5A1 mutation. Climacteric 2014, 17, 301–303. [Google Scholar] [CrossRef]
- Arafat, M.; Har-Vardi, I.; Harlev, A.; Levitas, E.; Zeadna, A.; Abofoul-Azab, M.; Dyomin, V.; Sheffield, V.C.; Lunenfeld, E.; Huleihel, M.; et al. Mutation in TDRD9 causes non-obstructive azoospermia in infertile men. J. Med. Genet. 2017, 54, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Pontoglio, A.; Bini, M. Two novel TEX15 mutations in a family with nonobstructive azoospermia. Gynecol. Obstet. Investig. 2017, 82, 283–286. [Google Scholar] [CrossRef]
- Okutman, O.; Muller, J.; Baert, Y.; Serdarogullari, M.; Gultomruk, M.; Piton, A.; Rombaut, C.; Benkhalifa, M.; Teletin, M.; Skory, V.; et al. Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum. Mol. Genet. 2015, 24, 5581–5588. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, L.; Yu, W.; Guo, H.; Zhang, H.; Wei, D.; Liang, L.; Feng, K.; Song, X.; Liu, Q.; et al. A novel functional variant in Wilms’ Tumor 1 (WT1) is associated with idiopathic non-obstructive azoospermia. Mol. Reprod. Dev. 2017, 84, 222–228. [Google Scholar] [CrossRef]
- Seabra, C.M.; Quental, S.; Lima, A.C.; Carvalho, F.; Gonçalves, J.; Fernandes, S.; Pereira, I.; Silva, J.; Marques, P.I.; Sousa, M.; et al. The mutational spectrum of WT1 in male infertility. J. Urol. 2015, 193, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Li, Z.S.; Ren, Y.; Jiang, T.; Wang, Y.Q.; Chen, M.; Zhang, J.; Hao, J.X.; Wang, Y.B.; Sha, R.N.; et al. The wilms tumor gene, Wt1, is critical for mouse spermatogenesis via regulation of sertoli cell polarity and is associated with non-obstructive azoospermia in humans. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum-Rakover, Y.; Weinberg-Shukron, A.; Renbaum, P.; Lobel, O.; Eideh, H.; Gulsuner, S.; Dahary, D.; Abu-Rayyan, A.; Kanaan, M.; Levy-Lahad, E.; et al. Minichromosome maintenance complex component 8 (MCM8) gene mutations result in primary gonadal failure. J. Med. Genet. 2015, 52, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Al-Agha, A.E.; Ahmed, I.A.; Nuebel, E.; Moriwaki, M.; Moore, B.; Peacock, K.A.; Mosbruger, T.; Neklason, D.W.; Jorde, L.B.; Yandell, M.; et al. Primary ovarian insufficiency and azoospermia in carriers of a homozygous PSMC3IP stop gain mutation. J. Clin. Endocrinol. Metab. 2018, 103, 555–563. [Google Scholar] [CrossRef]
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.S.; Lin, S.H.; Alper, S.L.; et al. Inhibition of WNK3 kinase signaling reduces brain damage and accelerates neurological recovery after stroke. Stroke 2015, 46, 1956–1965. [Google Scholar] [CrossRef]
- Nepal, M.; Che, R.; Zhang, J.; Ma, C.; Fei, P. Fanconi anemia signaling and cancer. Trends Cancer 2017, 3, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef]
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- Martin, C.A.; Ahmad, I.; Klingseisen, A.; Hussain, M.S.; Bicknell, L.S.; Leitch, A.; Nürnberg, G.; Toliat, M.R.; Murray, J.E.; Hunt, D.; et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat. Genet. 2014, 46, 1283–1292. [Google Scholar] [CrossRef]
- Harris, R.M.; Weiss, J.; Jameson, J.L. Male hypogonadism and germ cell loss caused by a mutation in Polo-like kinase 4. Endocrinology 2011, 152, 3975–3985. [Google Scholar] [CrossRef]
- Pace, A.J.; Lee, E.; Athirakul, K.; Coffman, T.M.; O’Brien, D.A.; Koller, B.H. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl− cotransporter. J. Clin. Investig. 2000, 105, 441–450. [Google Scholar] [CrossRef]
- Luo, M.; Yang, F.; Leu, N.A.; Landaiche, J.; Handel, M.A.; Benavente, R.; La Salle, S.; Wang, P.J. MEIOB exhibits single-stranded DNA-binding and exonuclease activities and is essential for meiotic recombination. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Souquet, B.; Abby, E.; Hervé, R.; Finsterbusch, F.; Tourpin, S.; Le Bouffant, R.; Duquenne, C.; Messiaen, S.; Martini, E.; Bernardino-Sgherri, J.; et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Caburet, S.; Todeschini, A.L.; Petrillo, C.; Martini, E.; Farran, N.D.; Legois, B.; Livera, G.; Younis, J.S.; Shalev, S.; Veitia, R.A. A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. EBioMedicine 2019, 42, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Miyado, M.; Saito, K.; Katsumi, M.; Nakamura, A.; Kobori, Y.; Tanaka, Y.; Ishikawa, H.; Yoshida, A.; Okada, H.; et al. Next-generation sequencing for patients with non-obstructive azoospermia: Implications for significant roles of monogenic/oligogenic mutations. Andrology 2017, 5, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.C.; Klur, S.; Watanabe, M.; Németh, A.H.; Le Ber, I.; Moniz, J.C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schöls, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Chen, J.; Wang, L.; Nie, L.; Long, J.; Chang, H.; Wu, J.; Huang, C.; Lei, M. The meiotic TERB1-TERB2-MAJIN complex tethers telomeres to the nuclear envelope. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef]
- Gómez-H., L.; Felipe-Medina, N.; Sánchez-Martín, M.; Davies, O.R.; Ramos, I.; García-Tuñón, I.; De Rooij, D.G.; Dereli, I.; Tóth, A.; Barbero, J.L.; et al. C14ORF39/SIX6OS1 is a constituent of the synaptonemal complex and is essential for mouse fertility. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Thorslund, T.; Esashi, F.; West, S.C. Interactions between human BRCA2 protein and the meiosis-specific recombinase DMC1. EMBO J. 2007, 26, 2915–2922. [Google Scholar] [CrossRef]
- Bishop, D.K.; Park, D.; Xu, L.; Kleckner, N. DMC1: A meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 1992, 69, 439–456. [Google Scholar] [CrossRef]
- Pittman, D.L.; Cobb, J.; Schimenti, K.J.; Wilson, L.A.; Cooper, D.M.; Brignull, E.; Handel, M.A.; Schimenti, J.C. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell 1998, 1, 697–705. [Google Scholar] [CrossRef]
- Du, L.; Luo, Y. Structure of a filament of stacked octamers of human DMC1 recombinase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Tock, A.J.; Henderson, I.R. Hotspots for initiation of meiotic recombination. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Bolcun-Filas, E.; Speed, R.; Taggart, M.; Grey, C.; De Massy, B.; Benavente, R.; Cooke, H.J. Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef]
- Horn, H.F.; Kim, D.I.; Wright, G.D.; Wong, E.S.M.; Stewart, C.L.; Burke, B.; Roux, K.J. A mammalian KASH domain protein coupling meiotic chromosomes to the cytoskeleton. J. Cell Biol. 2013, 202, 1023–1039. [Google Scholar] [CrossRef]
- Reynolds, A.; Qiao, H.; Yang, Y.; Chen, J.K.; Jackson, N.; Biswas, K.; Holloway, J.K.; Baudat, F.; De Massy, B.; Wang, J.; et al. RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nat. Genet. 2013, 45, 269–278. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Ogonuki, N.; Inou, K.; Ogura, A.; Handel, M.A.; Noguchi, J.; Kunieda, T. t-SNARE syntaxin2 (STX2) is implicated in intracellular transport of sulfoglycolipids during meiotic prophase in mouse spermatogenesis. Biol. Reprod. 2013, 88. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Li, H.Y.; He, W.B.; Tu, C.; Du, J.; Li, W.; Lu, G.X.; Lin, G.; Yang, Y.; Tan, Y.Q. XRCC2 mutation causes premature ovarian insufficiency as well as non-obstructive azoospermia in humans. Clin. Genet. 2019, 95, 442–443. [Google Scholar] [CrossRef]
- Herrán, Y.; Gutiérrez-Caballero, C.; Sánchez-Martín, M.; Hernández, T.; Viera, A.; Barbero, J.L.; De Álava, E.; De Rooij, D.G.; Suja, J.Á.; Llano, E.; et al. The cohesin subunit RAD21L functions in meiotic synapsis and exhibits sexual dimorphism in fertility. EMBO J. 2011, 30, 3091–3105. [Google Scholar] [CrossRef]
- Shibuya, H.; Ishiguro, K.I.; Watanabe, Y. The TRF1-binding protein TERB1 promotes chromosome movement and telomere rigidity in meiosis. Nat. Cell Biol. 2014, 16, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Hernández-Hernández, A.; Morimoto, A.; Negishi, L.; Höög, C.; Watanabe, Y. MAJIN links telomeric DNA to the nuclear membrane by exchanging telomere cap. Cell 2015, 163, 1252–1266. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Tolle, D.P.; Barrett, A.J. Evolutionary families of peptidase inhibitors. Biochem. J. 2004, 378, 705–716. [Google Scholar] [CrossRef]
- Tanaka, T.; Hosokawa, M.; Vagin, V.V.; Reuter, M.; Hayashi, E.; Mochizuki, A.L.; Kitamura, K.; Yamanaka, H.; Kondoh, G.; Okawa, K.; et al. Tudor domain containing 7 (Tdrd7) is essential for dynamic ribonucleoprotein (RNP) remodeling of chromatoid bodies during spermatogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10579–10584. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Si, Y.; Slaymaker, S.; Li, J.; Zheng, H.; Young, D.L.; Aslanian, A.; Saunders, L.; Verdin, E.; Charo, I.F. Zmynd15 encodes a histone deacetylase-dependent transcriptional repressor essential for spermiogenesis and male fertility. J. Biol. Chem. 2010, 285, 31418–31426. [Google Scholar] [CrossRef] [PubMed]
- Vinci, G.; Chantot-Bastaraud, S.; El Houate, B.; Lortat-Jacob, S.; Brauner, R.; McElreavey, K. Association of deletion 9p, 46,XY gonadal dysgenesis and autistic spectrum disorder. Mol. Hum. Reprod. 2007, 13, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A.; Nunes, M.; Quintana-Murci, L.; Rappaport, R.; Thibaud, E.; Jaubert, F.; Fellous, M.; McElreavey, K.; Gonçalves, J.; Silva, M.; et al. Swyer syndrome and 46,XY partial gonadal dysgenesis associated with 9p deletions in the absence of monosomy-9p syndrome. Am. J. Hum. Genet. 1998, 63, 901–905. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bogliolo, M.; Bluteau, D.; Lespinasse, J.; Pujol, R.; Vasquez, N.; D’Enghien, C.D.; Stoppa-Lyonnet, D.; Leblanc, T.; Soulier, J.; Surrallés, J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018, 20, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Catucci, I.; Osorio, A.; Arver, B.; Neidhardt, G.; Bogliolo, M.; Zanardi, F.; Riboni, M.; Minardi, S.; Pujol, R.; Azzollini, J.; et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. 2018, 20, 452–457. [Google Scholar] [CrossRef]
- Yan, Z.; Delannoy, M.; Ling, C.; Daee, D.; Osman, F.; Muniandy, P.A.; Shen, X.; Oostra, A.B.; Du, H.; Steltenpool, J.; et al. A Histone-fold complex and FANCM form a conserved dna-remodeling complex to maintain genome stability. Mol. Cell 2010, 37, 865–878. [Google Scholar] [CrossRef]
- Bakker, S.T.; van de Vrugt, H.J.; Rooimans, M.A.; Oostra, A.B.; Steltenpool, J.; Delzenne-Goette, E.; van der Wal, A.; van der Valk, M.; Joenje, H.; te Riele, H.; et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009, 18, 3484–3495. [Google Scholar] [CrossRef]
- Luo, Y.; Hartford, S.A.; Zeng, R.; Southard, T.L.; Shima, N.; Schimenti, J.C. Hypersensitivity of Primordial Germ Cells to Compromised Replication-Associated DNA Repair Involves ATM-p53-p21 Signaling. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Kato, Y.; Saga, Y. NANOS2 promotes male germ cell development independent of meiosis suppression. Dev. Biol. 2014, 385, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Sasaoka, Y.; Kiso, M.; Abe, K.; Haraguchi, S.; Kobayashi, S.; Saga, Y. Conserved role of nanos proteins in germ cell development. Science 2003, 301, 1239–1241. [Google Scholar] [CrossRef]
- Kusz-Zamelczyk, K.; Sajek, M.; Spik, A.; Glazar, R.; Jȩdrzejczak, P.; Latos-Bieleńska, A.; Kotecki, M.; Pawelczyk, L.; Jaruzelska, J. Mutations of NANOS1, a human homologue of the Drosophila morphogen, are associated with a lack of germ cells in testes or severe oligo-astheno-teratozoospermia. J. Med. Genet. 2013, 50, 187–193. [Google Scholar] [CrossRef]
- Greenbaum, M.P.; Iwamori, T.; Buchold, G.M.; Matzuk, M.M. Germ cell intercellular bridges. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–18. [Google Scholar] [CrossRef]
- Ruan, J.; He, X.J.; Du, W.D.; Chen, G.; Zhou, Y.; Xu, S.; Zuo, X.B.; Fang, L.B.; Cao, Y.X.; Zhang, X.J. Genetic variants in TEX15 gene conferred susceptibility to spermatogenic failure in the Chinese Han population. Reprod. Sci. 2012, 19, 1190–1196. [Google Scholar] [CrossRef]
- Nachtigal, M.W.; Hirokawa, Y.; Enyeart-VanHouten, D.L.; Flanagan, J.N.; Hammer, G.D.; Ingraham, H.A. Wilms’ tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell 1998, 93, 445–454. [Google Scholar] [CrossRef]
- Cools, M.; Nordenström, A.; Robeva, R.; Hall, J.; Westerveld, P.; Flück, C.; Köhler, B.; Berra, M.; Springer, A.; Schweizer, K.; et al. Caring for individuals with a difference of sex development (DSD): A Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 1–21. [Google Scholar] [CrossRef]
- Ferraz-de-Souza, B.; Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol. Cell. Endocrinol. 2011, 336, 198–205. [Google Scholar] [CrossRef]
- Hastie, N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Okita, H.; Haruta, M.; Arai, Y.; Oue, T.; Tanaka, Y.; Horie, H.; Hinotsu, S.; Koshinaga, T.; Yoneda, A.; et al. A high incidence of WT1 abnormality in bilateral Wilms tumours in Japan, and the penetrance rates in children with WT1 germline mutation. Br. J. Cancer 2015, 112, 1121–1133. [Google Scholar] [CrossRef]
- Lourenço, D.; Brauner, R.; Lin, L.; De Perdigo, A.; Weryha, G.; Muresan, M.; Boudjenah, R.; Guerra-Junior, G.; Maciel-Guerra, A.T.; Achermann, J.C.; et al. Mutations in NR5A1 associated with ovarian insufficiency. N. Engl. J. Med. 2009, 360, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Köhler, B.; Biebermann, H.; Friedsam, V.; Gellermann, J.; Maier, R.F.; Pohl, M.; Wieacker, P.; Hiort, O.; Grüters, A.; Krude, H. Analysis of the Wilms’ tumor suppressor gene (WT1) in patients 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 2011, 96. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shoji, M.; Tanaka, T.; Hosokawa, M.; Reuter, M.; Stark, A.; Kato, Y.; Kondoh, G.; Okawa, K.; Chujo, T.; Suzuki, T.; et al. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev. Cell 2009, 17, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.C.; Trakselis, M.A. The MCM8/9 complex: A recent recruit to the roster of helicases involved in genome maintenance. DNA Repair 2019, 76, 1–10. [Google Scholar] [CrossRef]
- Zangen, D.; Kaufman, Y.; Zeligson, S.; Perlberg, S.; Fridman, H.; Kanaan, M.; Abdulhadi-Atwan, M.; Abu Libdeh, A.; Gussow, A.; Kisslov, I.; et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 2011, 89, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Sung, P. Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res. 2015, 43, 4055–4066. [Google Scholar] [CrossRef]
- Sansam, C.L.; Pezza, R.J. Connecting by breaking and repairing: Mechanisms of DNA strand exchange in meiotic recombination. FEBS J. 2015, 282, 2431–2444. [Google Scholar] [CrossRef]
- Petukhova, G.V.; Romanienko, P.J.; Camerini-Otero, R.D. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev. Cell 2003, 5, 927–936. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Hovatta, O.; La Marca, A.; Livera, G.; Monniaux, D.; Persani, L.; Heddar, A.; Jarzabek, K.; Laisk-Podar, T.; Salumets, A.; et al. Advances in the molecular pathophysiology, genetics, and treatment of primary ovarian insufficiency. Trends Endocrinol. Metab. 2018, 29, 400–419. [Google Scholar] [CrossRef]
- Laissue, P. Aetiological coding sequence variants in non-syndromic premature ovarian failure: From genetic linkage analysis to next generation sequencing. Mol. Cell. Endocrinol. 2015, 411, 243–257. [Google Scholar] [CrossRef]
- Rossetti, R.; Ferrari, I.; Bonomi, M.; Persani, L. Genetics of primary ovarian insufficiency. Clin. Genet. 2017, 91, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zhang, H.; Ke, H.; Zhang, J.; Cheng, L.; Liu, Y.; Qin, Y.; Chen, Z.J. Premature ovarian insufficiency: Phenotypic characterization within different etiologies. J. Clin. Endocrinol. Metab. 2017, 102, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Golezar, S.; Ramezani Tehrani, F.; Khazaei, S.; Ebadi, A.; Keshavarz, Z. The global prevalence of primary ovarian insufficiency and early menopause: A meta-analysis. Climacteric 2019, 22, 403–411. [Google Scholar] [CrossRef]
- Patiño, L.C.; Beau, I.; Carlosama, C.; Buitrago, J.C.; González, R.; Suárez, C.F.; Patarroyo, M.A.; Delemer, B.; Young, J.; Binart, N.; et al. New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum. Reprod. 2017, 32, 1512–1520. [Google Scholar] [CrossRef]
- Fouquet, B.; Pawlikowska, P.; Caburet, S.; Guigon, C.; Mäkinen, M.; Tanner, L.; Hietala, M.; Urbanska, K.; Bellutti, L.; Legois, B.; et al. A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Jiao, J.; Zhang, F.; Pan, Y.; Wang, Q.; Chen, Q.; Cai, B.; Tang, S.; Zhou, Z.; et al. Rare variants in FANCA induce premature ovarian insufficiency. Hum. Genet. 2019, 138, 1227–1236. [Google Scholar] [CrossRef]
- Heddar, A.; Misrahi, M. Concerns regarding the potentially causal role of FANCA heterozygous variants in human primary ovarian insufficiency. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- AlAsiri, S.; Basit, S.; Wood-Trageser, M.A.; Yatsenko, S.A.; Jeffries, E.P.; Surti, U.; Ketterer, D.M.; Afzal, S.; Ramzan, K.; Faiyaz-Ul Haque, M.; et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Investig. 2015, 125, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Guo, T.; Li, G.; Zhou, L.G.; Qin, Y.; Chen, Z.J. Minichromosome maintenance complex component 8 mutations cause primary ovarian insufficiency. Fertil. Steril. 2016, 106, 1485–1489.e2. [Google Scholar] [CrossRef] [PubMed]
- Bouali, N.; Francou, B.; Bouligand, J.; Imanci, D.; Dimassi, S.; Tosca, L.; Zaouali, M.; Mougou, S.; Young, J.; Saad, A.; et al. New MCM8 mutation associated with premature ovarian insufficiency and chromosomal instability in a highly consanguineous Tunisian family. Fertil. Steril. 2017, 108, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Heddar, A.; Beckers, D.; Fouquet, B.; Roland, D.; Misrahi, M. A novel phenotype combining primary ovarian insufficiency growth retardation and pilomatricomas with MCM8 mutation. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, S.; Li, P. Two novel mutations in the MCM8 gene shared by two Chinese siblings with primary ovarian insufficiency and short stature. Mol. Genet. Genomic Med. 2020, 8. [Google Scholar] [CrossRef]
- Zhang, Y.X.; He, W.B.; Xiao, W.J.; Meng, L.L.; Tan, C.; Du, J.; Lu, G.X.; Lin, G.; Tan, Y.Q. Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol. Genet. Genomic Med. 2020, 8. [Google Scholar] [CrossRef]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef]
- Caburet, S.; Arboleda, V.A.; Llano, E.; Overbeek, P.A.; Barbero, J.L.; Oka, K.; Harrison, W.; Vaiman, D.; Ben-Neriah, Z.; García-Tuñón, I.; et al. Mutant cohesin in premature ovarian failure. N. Engl. J. Med. 2014, 370, 943–949. [Google Scholar] [CrossRef]
- Le Quesne Stabej, P.; Williams, H.J.; James, C.; Tekman, M.; Stanescu, H.C.; Kleta, R.; Ocaka, L.; Lescai, F.; Storr, H.L.; Bitner-Glindzicz, M.; et al. STAG3 truncating variant as the cause of primary ovarian insufficiency. Eur. J. Hum. Genet. 2016, 24, 135–138. [Google Scholar] [CrossRef]
- Colombo, R.; Pontoglio, A.; Bini, M. A STAG3 missense mutation in two sisters with primary ovarian insufficiency. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 216, 269–271. [Google Scholar] [CrossRef]
- He, W.B.; Banerjee, S.; Meng, L.L.; Du, J.; Gong, F.; Huang, H.; Zhang, X.X.; Wang, Y.Y.; Lu, G.X.; Lin, G.; et al. Whole-exome sequencing identifies a homozygous donor splice-site mutation in STAG3 that causes primary ovarian insufficiency. Clin. Genet. 2018, 93, 340–344. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Nishi, M.Y.; Funari, M.F.A.; Lerario, A.M.; Baracat, E.C.; Hayashida, S.A.Y.; Maciel, G.A.R.; Jorge, A.A.L.; Mendonca, B.B. Two rare loss-of-function variants in the STAG3 gene leading to primary ovarian insufficiency. Eur. J. Med. Genet. 2019, 62, 186–189. [Google Scholar] [CrossRef]
- Heddar, A.; Dessen, P.; Flatters, D.; Misrahi, M. Novel STAG3 mutations in a Caucasian family with primary ovarian insufficiency. Mol. Genet. Genomics 2019, 294, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.J.; He, W.B.; Zhang, Y.X.; Meng, L.L.; Lu, G.X.; Lin, G.; Tan, Y.Q.; Du, J. In-Frame Variants in STAG3 gene cause premature ovarian insufficiency. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.; Hwang, G.; Jacob, J.; Sapp, N.; Bedigian, R.; Oka, K.; Overbeek, P.; Murray, S.; Jordan, P.W. Meiosis-specific cohesin component, stag3 is essential for maintaining centromere chromatid cohesion, and required for DNA repair and synapsis between homologous chromosomes. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.; Behar, D.M.; Smirin-Yosef, P.; Lagovsky, I.; Tzur, S.; Basel-Vanagaite, L. Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 2014, 99, E2129–E2132. [Google Scholar] [CrossRef] [PubMed]
- Hernández-López, D.; Geisinger, A.; Trovero, M.F.; Santiñaque, F.F.; Brauer, M.; Folle, G.A.; Benavente, R.; Rodríguez-Casuriaga, R. Familial primary ovarian insufficiency associated with an SYCE1 point mutation: Defective meiosis elucidated in humanized mice. Mol. Hum. Reprod. 2020, 26, 485–497. [Google Scholar] [CrossRef]
- Zhe, J.; Ye, D.; Chen, X.; Liu, Y.; Zhou, X.; Li, Y.; Zhang, J.; Chen, S. Consanguineous Chinese familial study reveals that a gross deletion that includes the SYCE1 gene region is associated with premature ovarian insufficiency. Reprod. Sci. 2020, 27, 461–467. [Google Scholar] [CrossRef]
- Carlosama, C.; Elzaiat, M.; Patiño, L.C.; Mateus, H.E.; Veitia, R.A.; Laissue, P. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum. Mol. Genet. 2017, 26, 3161–3166. [Google Scholar] [CrossRef]
- Kneitz, B.; Cohen, P.E.; Avdievich, E.; Zhu, L.; Kane, M.F.; Hou, H.; Kolodner, R.D.; Kucherlapati, R.; Pollard, J.W.; Edelmann, W. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000, 14, 1085–1097. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ridgeway, A.D.; Lamb, D.J. DNA mismatch repair and infertility. Curr. Opin. Urol. 2010, 20, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Janse, F.; De With, L.M.; Duran, K.J.; Kloosterman, W.P.; Goverde, A.J.; Lambalk, C.B.; Laven, J.S.E.; Fauser, B.C.J.M.; Giltay, J.C. Limited contribution of NR5A1 (SF-1) mutations in women with primary ovarian insufficiency (POI). Fertil. Steril. 2012, 97. [Google Scholar] [CrossRef] [PubMed]
- Adamovic, T.; Chen, Y.; Thai, H.T.T.; Zhang, X.; Markljung, E.; Zhao, S.; Nordenskjöld, A. The p.G146A and p.P125P polymorphisms in the steroidogenic factor-1 (SF-1) gene do not affect the risk for hypospadias in caucasians. Sex. Dev. 2012, 6, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Dati, E.; Baldinotti, F.; Toschi, B.; Marrocco, G.; Sessa, M.R.; Michelucci, A.; Simi, P.; Baroncelli, G.I. NR5A1 gene mutations: Clinical, endocrine and genetic features in two girls with 46,XY disorder of sex development. Horm. Res. Paediatr. 2014, 81, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Köhler, B.; Lin, L.; Ferraz-de-Souza, B.; Wieacker, P.; Heidemann, P.; Schröder, V.; Biebermann, H.; Schnabel, D.; Grüters, A.; Achermann, J.C. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum. Mutat. 2008, 29, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Philibert, P.; Paris, F.; Lakhal, B.; Audran, F.; Gaspari, L.; Saâd, A.; Christin-Maître, S.; Bouchard, P.; Sultan, C. NR5A1 (SF-1) gene variants in a group of 26 young women with XX primary ovarian insufficiency. Fertil. Steril. 2013, 99, 484–489.e1. [Google Scholar] [CrossRef]
- Voican, A.; Bachelot, A.; Bouligand, J.; Francou, B.; Dulon, J.; Lombès, M.; Touraine, P.; Guiochon-Mantel, A. NR5A1 (SF-1) mutations are not a major cause of primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 2013, 98. [Google Scholar] [CrossRef]
- Bertrand-Delepine, J.; Manouvrier-hanu, S.; Cartigny, M.; Paris, F.; Mallet, D.; Philibert, P.; Morel, Y.; Lefevre, C.; Dewailly, D.; Catteau-jonard, S. In cases of familial primary ovarian insufficiency and disorders of gonadal development, consider NR5A1/SF-1 sequence variants. Reprod. Biomed. Online 2020, 40, 151–159. [Google Scholar] [CrossRef]
- Lynch, D.R.; Braastad, C.D.; Nagan, N. Ovarian failure in ataxia with oculomotor apraxia type 2. Am. J. Med. Genet. Part A 2007, 143, 1775–1777. [Google Scholar] [CrossRef]
- Pérez de Nanclares, G.; Castaño, L.; Bilbao, J.R.; Vallo, A.; Rica, I.; Vela, A.; Martul, P. Molecular analysis of Frasier syndrome: Mutation in the WT1 gene in a girl with gonadal dysgenesis and nephronophthisis. J. Pediatr. Endocrinol. Metab. 2002, 15, 1047–1050. [Google Scholar] [CrossRef]
- Barbaux, S.; Niaudet, P.; Gubler, M.C.; Grünfeld, J.P.; Jaubert, F.; Kuttenn, F.; Fékété, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.-W.; Sha, Y.-K.; Ji, Z.-Y.; Mei, L.-B.; Ding, L.; Zhang, Q.; Qiu, P.-P.; Lin, S.-B.; Wang, X.; Li, P.; et al. TSGA10 is a novel candidate gene associated with acephalic spermatozoa. Clin. Genet. 2017, 93, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, G.; Zhang, J.; Gao, F.; Li, W.; Qin, Y.; Chen, Z.J. Novel WT1 missense mutations in han Chinese women with premature ovarian failure. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C. Editorial for the special issue on the molecular genetics of male infertility. Hum. Genet. 2021, 140, 1–5. [Google Scholar] [CrossRef]
- Houston, B.J.; Conrad, D.F.; O’Bryan, M.K. A framework for high-resolution phenotyping of candidate male infertility mutants: From human to mouse. Hum. Genet. 2020, 140, 1–28. [Google Scholar] [CrossRef]
- Xavier, M.J.; Salas-Huetos, A.; Oud, M.S.; Aston, K.I.; Veltman, J.A. Disease gene discovery in male infertility: Past, present and future. Hum. Genet. 2021, 140, 7–19. [Google Scholar] [CrossRef]
- Capalbo, A.; Poli, M.; Riera-Escamilla, A.; Shukla, V.; Kudo Høffding, M.; Krausz, C.; Hoffmann, E.R.; Simon, C. Preconception genome medicine: Current state and future perspectives to improve infertility diagnosis and reproductive and health outcomes based on individual genomic data. Hum. Reprod. Update 2021, 27, 254–279. [Google Scholar] [CrossRef]
- Yadav, R.P.; Kotaja, N. Small RNAs in spermatogenesis. Mol. Cell Endocrinol. 2014, 382, 498–508. [Google Scholar] [CrossRef]
- Bo, H.; Liu, Z.; Zhu, F.; Zhou, D.; Tan, Y.; Zhu, W.; Fan, L. Long noncoding RNAs expression profile and long noncoding RNA-mediated competing endogenous RNA network in nonobstructive azoospermia patients. Epigenomics 2020, 12, 673–684. [Google Scholar] [CrossRef]
- Gunes, S.; Esteves, S.C. Role of genetics and epigenetics in male infertility. Andrologia 2021, 53, e13586. [Google Scholar] [CrossRef]
- Uysal, F.; Akkoyunlu, G.; Ozturk, S. Decreased expression of DNA methyltransferases in the testes of patients with non-obstructive azoospermia leads to changes in global DNA methylation levels. Reprod. Fertil. Dev. 2019. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| (a) | |||||||||||
| Gene ^ | OMIM | Locus ° | Function + | Inheritance | Other Phenotypes | POI | Mouse Reproductive Phenotypes # | Segregation in Family | More than One Unrelated Carrier | Independent Cohorts | Refs. |
| FANCA | 607139 | 16q24.3 | Interstrand crosslink repair | AR | Fanconi Anemia | Yes | Abnormal male meiosis, decreased germ cell number, decreased mature ovarian follicle number, absent ovarian follicles | Yes | Yes | No | [82] |
| PLK4 | 605031 | 4q28.1 | Centriole duplication during the cell cycle | AD | Microcephaly and chorioretinopathy | No | Decreased male germ cell number | No | No | No | [83] |
| WNK3 | 300358 | Xp11.22 | Regulation of electrolyte homeostasis, cell signaling, survival and proliferation | XLR | n.r. | No | Normal * | Yes | No | No | [84] |
| (b) | |||||||||||
| Gene ^ | OMIM | Locus ° | Function + | Inheritance | Other Phenotypes | POI | Mouse Reproductive Phenotypes # | Segregation in Family | More than One Unrelated Carrier | Independent Cohorts | Refs. |
| ADAD2 | n.a. | 16q24.1 | dsRNA-binding protein, RNA editing | AR | n.r. | No | Male and female infertility | Yes | Yes | Yes | [10] |
| C14orf39 | 617307 | 14q23.1 | Chromosome synapsis during meiotic recombination | AR | n.r | Yes | Arrest of male meiosis, abnormal chiasmata formation, abnormal chromosomal synapsis, abnormal X-Y chromosome synapsis during male meiosis, absent oocytes | Yes ** | Yes | No | [85] |
| DMC1 | 602721 | 22q13.1 | Meiotic recombination, DNA DSB repair | AR | n.r. | Yes | Arrest of male meiosis decreased oocyte number, absent oocytes, absent ovarian follicles, abnormal female meiosis | Yes ** | No | No | [86] |
| KASH5 | 618125 | 19q13.33 | Meiotic telomere attachment to nuclear envelope in the prophase of meiosis, homolog pairing during meiotic prophase | AR | n.r. | No | Arrest of male meiosis, female infertility | Yes | No | No | [84] |
| MAJIN | 617130 | 11q13.1 | Meiotic telomere attachment to the nucleus inner membrane during homologous pairing and synapsis | AR | n.r. | No | Meiotic arrest, male and female infertility | No | No | No | [87] |
| MEI1 | 608797 | 22q13.2 | Meiotic chromosome synapsis, DBS formation | AR | Hydatidiform mole | Yes | Arrest of male meiosis, female infertility | Yes | Yes | Yes | [10,88,89] |
| MEIOB | 617670 | 16p13.3 | DNA DSB repair, crossover formation and promotion to complete synapsis | AR | n.r. | Yes | Arrest of spermatogenesis, decreased oocyte number, absent oocytes | Yes | Yes | Yes | [10,90,91] |
| MSH4 | 602105 | 1p31.1 | Homologous chromosomes recombination and segregation at meiosis I | AR | n.r. | Yes | Azoospermia, abnormal male and female meiosis | No | Yes | Yes | [10] |
| RAD21L1 | n.a. | 20p13 | Meiosis-specific component of some cohesin complex | AR | n.r. | No | Arrest of male meiosis, absent oocytes, decreased mature ovarian follicle number, absent primordial ovarian follicles | Yes | No | No | [10] |
| RNF212 | 612041 | 4p16.3 | Regulator of crossing-over during meiosis | AR | n.r. | No | Arrest of male meiosis, female infertility | Yes | No | No | [92] |
| SETX | 608465 | 9q34.13 | DNA and RNA processing | AR | Amyotrophic lateral sclerosis; ataxia with oculomotor apraxia type 2 | Yes | Arrest of male meiosis, globozoospermia, reduced female fertility | No | Yes | Yes | [93,94] |
| SHOC1 | 618038 | 9q31.3 | Binds to single-stranded DNA and DNA branched structures; formation of crossover recombination intermediates | AR | n.r. | No | Arrest of male meiosis | Yes | Yes | Yes | [10,95] |
| SPINK2 | 605753 | 4q12 | Inhibitor of acrosin | AR | n.r. | No | Kinked sperm flagellum, oligozoospermia, teratozoospermia, abnormal male germ cell apoptosis | Yes | No | No | [1,96] |
| SPO11 | 605114 | 20q13.31 | Initiation of DSBs | AR | n.r. | No | Arrest of male meiosis, decreased oocyte number, oocyte degeneration, abnormal female meiosis | Yes | No | No | [84] |
| STAG3 | 608489 | 7q22.1 | Cohesion of sister chromatids, DNA DSB repair | AR | n.r. | Yes | Azoospermia, absent oocytes | Yes ** | Yes | Yes | [10,92,97,98] |
| STX2 | 132350 | 12q24.33 | Sulfoglycolipid transporter | AR | n.r. | No | Arrest of male meiosis | No | No | No | [99] |
| SYCE1 | 611486 | 10q26.3 | Chromosome synapsis in meiosis | AR | n.r. | Yes | Arrest of male meiosis, decreased mature ovarian follicle number | Yes | Yes | Yes | [10,100,101] |
| TDRD7 | 611258 | 9q22.33 | RNA processing | AR | Congenital cataract | No | Arrest of spermatogenesis, abnormal male germ cell apoptosis | Yes | Yes | No | [2,102] |
| TERB1 | 617332 | 16q22.1 | Meiotic telomere attachment to the nucleus inner membrane during homologous pairing and synapsis | AR | n.r. | No | Arrest of male meiosis, absent oocytes, absent ovarian follicles, abnormal female meiosis I arrest | Yes | Yes | Yes | [10,87] |
| TERB2 | 617131 | 15q21.1 | Meiotic telomere attachment to the nucleus inner membrane during homologous pairing and synapsis | AR | n.r. | No | Arrest of male meiosis, absent ovarian follicles, abnormal female meiosis | Yes | No | No | [87] |
| TEX11 | 300311 | Xq13.1 | Chromosome synapsis and formation of crossovers | XLR | n.r. | No | Arrest of male meiosis, meiotic non-disjunction during M1 phase | Yes | Yes | Yes | [10,103,104,105] |
| XRCC2 | 600375 | 7q36.1 | Interstrand crosslink repair, DNA DSB repair | AR | Fanconi Anemia | Yes | Meiotic arrest, POI | Yes ** | No | No | [106] |
| ZMYND15 | 614312 | 17p13.2 | Transcriptional repressor | AR | n.r. | No | Azoospermia | Yes | No | No | [107] |
| (c) | |||||||||||
| Gene ^ | OMIM | Locus ° | Function + | Inheritance | Other Phenotypes | POI | Mouse Reproductive Phenotypes # | Segregation in Family | More than One Unrelated Carrier | Independent Cohorts | Refs. |
| DMRT1 | 602424 | 9p24.3 | Transcription factor involved in male sex determination and differentiation | AD | Ambiguous genitalia and sex reversal | No | Abnormal male meiosis, male infertility | Yes | Yes | Yes | [10,108] |
| FANCM | 609644 | 14q21.2 | DNA DSB repair, interstrand cross-link removal | AR | n.r. | Yes | Azoospermia, decreased mature ovarian follicle number | Yes | Yes | Yes | [109,110] |
| M1AP | 619098 | 2p13.1 | Meiosis I progression | AR | n.r. | No | From arrest of male meiosis to severe oligozoospermia/globozoospermia | Yes | Yes | Yes | [111] |
| NANOS2 | 608228 | 19q13.32 | Spermatogonial stem cell maintenance | AR | n.r. | No | Azoospermia, abnormal female meiosis | Yes | Yes | No | [84] |
| NR5A1 | 184757 | 9q33.3 | transcriptional activator for sex determination | AD | 46, XY and 46, XX sex reversal; adrenocortical insufficiency | Yes | From oligozoospermia to arrest of spermatogenesis, decreased mature ovarian follicle number, absent mature ovarian follicles | Yes ** | Yes | Yes | [112,113,114,115,116] |
| TAF4B | 601689 | 18q11.2 | Transcriptional coactivator | AR | n.r. | No | Oligozoospermia, decreased male germ cell number, asthenozoospermia, absent mature ovarian follicles, impaired ovarian folliculogenesis | Yes | No | No | [107] |
| TDRD9 | 617963 | 14q32.33 | Repression of transposable elements during meiosis | AR | n.r. | No | Arrest of male meiosis | Yes | No | No | [117] |
| TEX14 | 605792 | 17q22 | Formation of meiotic intercellular bridges | AR | n.r. | No | Arrest of male meiosis | Yes | Yes | Yes | [10,84,90] |
| TEX15 | 605795 | 8p12 | Chromosome, synapsis, DNA DSB repair | AR | n.r. | No | Arrest of male meiosis | Yes | Yes | Yes | [118,119] |
| WT1 | 607102 | 11p13 | Transcription factor | AD | Wilms tumor type 1; Nephrotic sdr type 4; Denys-Drash sdr; Frasier sdr; Meacham sdr; Mesothelioma | Yes | Azoospermia | No | Yes | Yes | [120,121,122] |
| (d) | |||||||||||
| Gene ^ | OMIM | Locus ° | Function + | Inheritance | Other Phenotypes | POI | Mouse Reproductive Phenotypes # | Segregation in Family | More than One Unrelated Carrier | Independent Cohorts | Refs. |
| MCM8 | 608187 | 20p12.3 | DNA DSB repair, interstrand crosslink removal | AR | n.r. | Yes | Arrest of male meiosis, decreased oocyte number, decreased mature ovarian follicle number, increased ovary tumor incidence, increased ovary adenoma incidence | Yes ** | No | No | [123] |
| PSMC3IP | 608665 | 17q21.2 | Stimulating DMC1-mediated strand exchange required for pairing homologous chromosomes during meiosis. | AR | Ovarian dysgenesis | Yes | Arrest of male meiosis, absent ovarian follicles, abnormal ovary development | Yes ** | No | No | [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioppi, F.; Rosta, V.; Krausz, C. Genetics of Azoospermia. Int. J. Mol. Sci. 2021, 22, 3264. https://doi.org/10.3390/ijms22063264
Cioppi F, Rosta V, Krausz C. Genetics of Azoospermia. International Journal of Molecular Sciences. 2021; 22(6):3264. https://doi.org/10.3390/ijms22063264
Chicago/Turabian StyleCioppi, Francesca, Viktoria Rosta, and Csilla Krausz. 2021. "Genetics of Azoospermia" International Journal of Molecular Sciences 22, no. 6: 3264. https://doi.org/10.3390/ijms22063264
APA StyleCioppi, F., Rosta, V., & Krausz, C. (2021). Genetics of Azoospermia. International Journal of Molecular Sciences, 22(6), 3264. https://doi.org/10.3390/ijms22063264