
NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle

 , , , and
, , , and 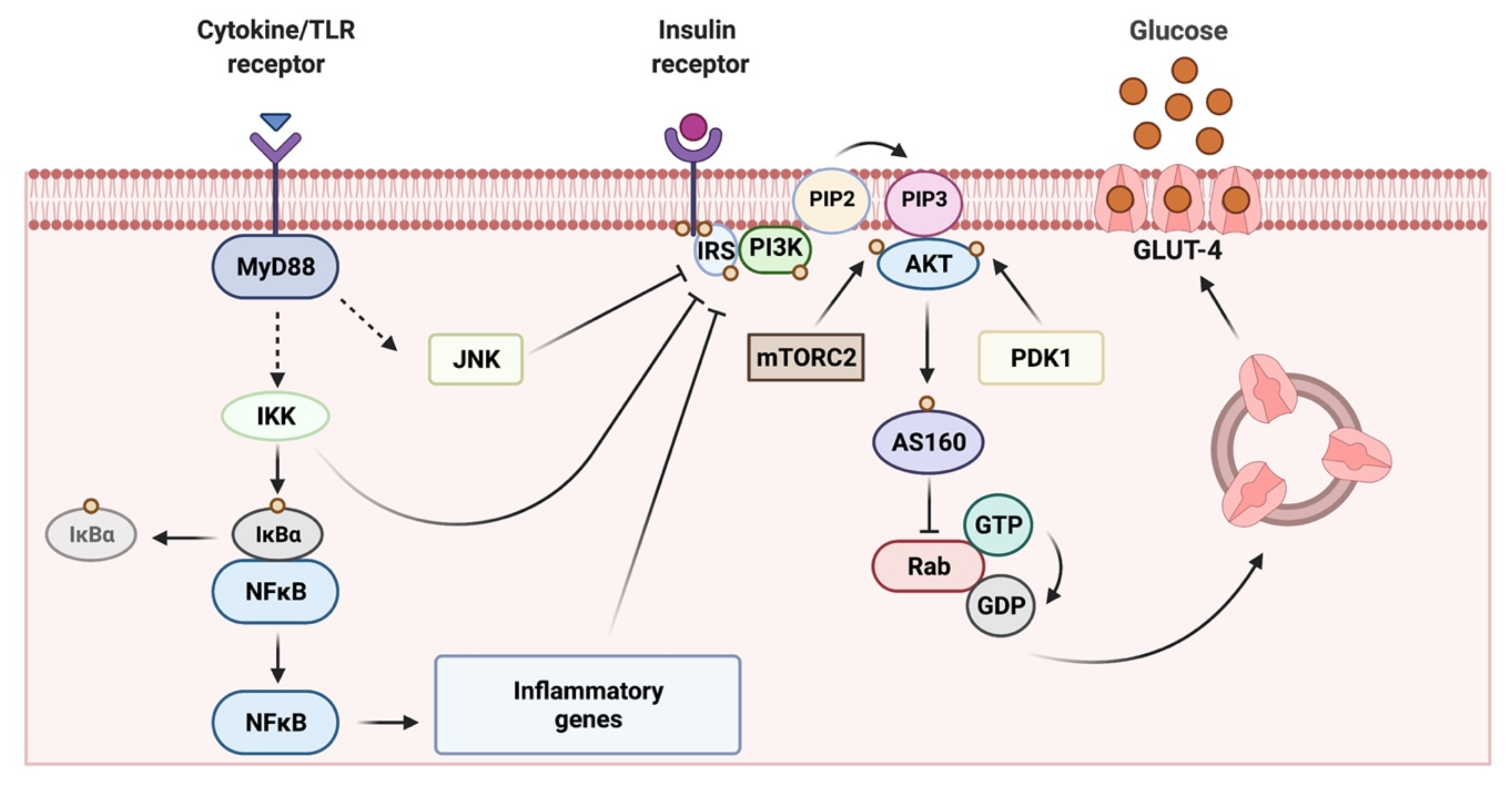{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Skeletal Muscle as a Key Target Organ for Insulin Actions
3. Obesity-Related Chronic Low-Grade Inflammation in Skeletal Muscle and Its Contribution to Insulin Resistance
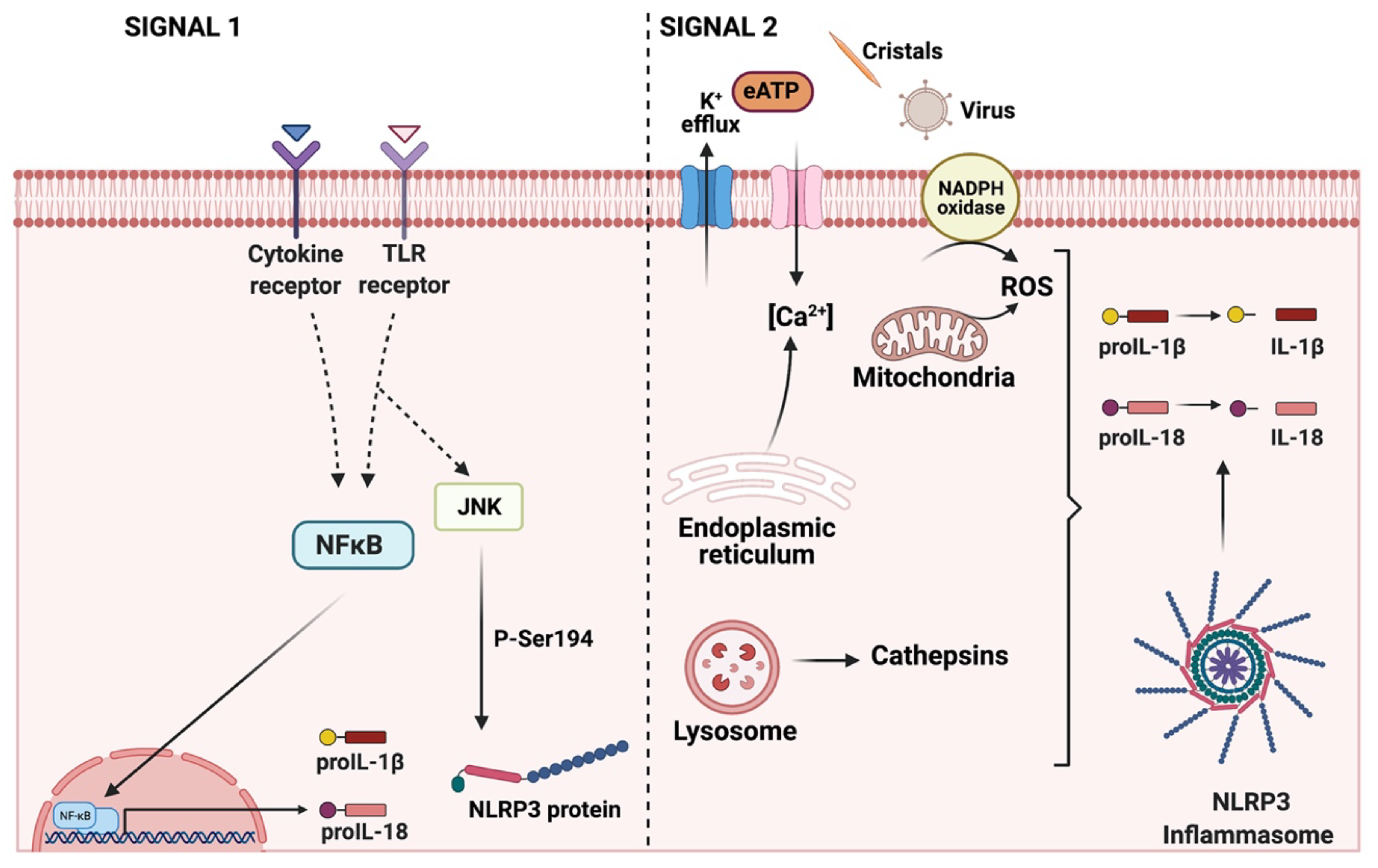
4. NLRP3 Inflammasome: Mechanisms of Activation and Downstream Effectors
4.1. ROS-Mediated NLRP3 Inflammasome Activation
4.2. Ionic-Mediated Pathways for NLRP3 Inflammasome Activation
4.3. Lysosomal Dysfunction-Mediated NLRP3 Inflammasome Activation
5. Gasdermins: A Specific Pathway for IL-1β Secretion
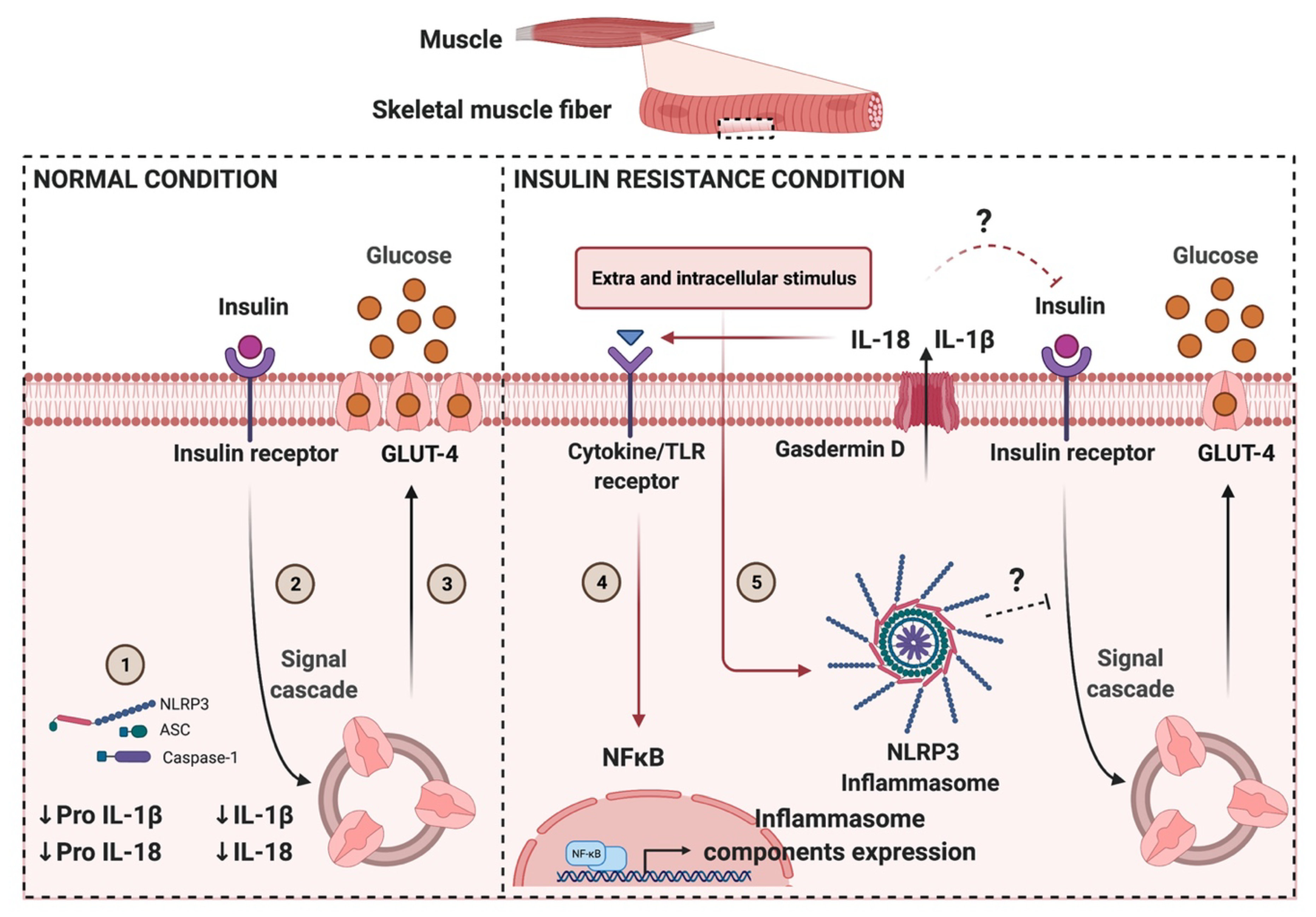
6. NLRP3 Role in Metabolic Disorders: Limited Information in Skeletal Muscle
7. Skeletal Muscle Lipid Infiltration: Possible Role on Local Inflammation?
8. NLRP3 Inflammasome Inhibitors
9. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Ferrannini, E.; Smith, J.D.; Cobelli, C.; Toffolo, G.; Pilo, A.; DeFronzo, R.A. Effect of insulin on the distribution and disposition of glucose in man. J. Clin. Investig. 1985, 76, 357–364. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Gunnarsson, R.; Björkman, O.; Olsson, M.; Wahren, J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J. Clin. Investig. 1985, 76, 149–155. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; Sun, Y.; Chiu, T.T.; Foley, K.P. Signal transduction meets vesicle traffic: The software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 2014, 306, C879–C886. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Zierath, J.R.; Kane, S.; Krook, A.; Lienhard, G.E.; Wallberg-Henriksson, H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 2005, 54, 1692–1697. [Google Scholar] [CrossRef]
- Lauritzen, H.P.; Ploug, T.; Prats, C.; Tavaré, J.M.; Galbo, H. Imaging of insulin signaling in skeletal muscle of living mice shows major role of T-tubules. Diabetes 2006, 55, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hansen, P.A.; Marshall, B.A.; Holloszy, J.O.; Mueckler, M. Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J. Cell Biol. 1996, 135, 415–430. [Google Scholar] [CrossRef]
- Lauritzen, H.P.; Galbo, H.; Brandauer, J.; Goodyear, L.J.; Ploug, T. Large GLUT4 vesicles are stationary while locally and reversibly depleted during transient insulin stimulation of skeletal muscle of living mice: Imaging analysis of GLUT4-enhanced green fluorescent protein vesicle dynamics. Diabetes 2008, 57, 315–324. [Google Scholar] [CrossRef]
- Rose, A.J.; Richter, E.A. Skeletal muscle glucose uptake during exercise: How is it regulated? Physiology 2005, 20, 260–270. [Google Scholar] [CrossRef]
- Love, D.C.; Hanover, J.A. The hexosamine signaling pathway: Deciphering the “O-GlcNAc code”. Sci. Stke 2005, 2005, re13. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Kauffman, F.C.; Max, S.R. The pentose phosphate pathway in regenerating skeletal muscle. Biochem. J. 1978, 170, 17–22. [Google Scholar] [CrossRef]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Zierath, J.R.; He, L.; Gumà, A.; Odegoard Wahlström, E.; Klip, A.; Wallberg-Henriksson, H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 1996, 39, 1180–1189. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Sinacore, D.R.; Gulve, E.A. The role of skeletal muscle in glucose transport, glucose homeostasis, and insulin resistance: Implications for physical therapy. Phys. Ther. 1993, 73, 878–891. [Google Scholar] [CrossRef]
- Fink, L.N.; Costford, S.R.; Lee, Y.S.; Jensen, T.E.; Bilan, P.J.; Oberbach, A.; Blüher, M.; Olefsky, J.M.; Sams, A.; Klip, A. Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity 2014, 22, 747–757. [Google Scholar] [CrossRef]
- Khan, I.M.; Perrard, X.Y.; Brunner, G.; Lui, H.; Sparks, L.M.; Smith, S.R.; Wang, X.; Shi, Z.Z.; Lewis, D.E.; Wu, H.; et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int. J. Obes. 2015, 39, 1607–1618. [Google Scholar] [CrossRef]
- Fink, L.N.; Oberbach, A.; Costford, S.R.; Chan, K.L.; Sams, A.; Blüher, M.; Klip, A. Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia 2013, 56, 1623–1628. [Google Scholar] [CrossRef]
- Green, C.J.; Pedersen, M.; Pedersen, B.K.; Scheele, C. Elevated NF-κB activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by AMP-activated protein kinase. Diabetes 2011, 60, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Ryan, A.J.; Mudaliar, S.R.; Henry, R.R. Altered Myokine Secretion Is an Intrinsic Property of Skeletal Muscle in Type 2 Diabetes. PLoS ONE 2016, 11, e0158209. [Google Scholar] [CrossRef] [PubMed]
- Jorquera, G.; Meneses-Valdés, R.; Rosales-Soto, G.; Valladares-Ide, D.; Campos, C.; Silva-Monasterio, M.; Llanos, P.; Cruz, G.; Jaimovich, E.; Casas, M. High extracellular ATP levels released through pannexin-1 channels mediate inflammation and insulin resistance in skeletal muscle fibers of diet induced obese mice. Diabetologia 2021. accepted for publication. [Google Scholar] [CrossRef]
- Ye, J. Mechanisms of insulin resistance in obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Sabio, G.; Kennedy, N.J.; Cavanagh-Kyros, J.; Jung, D.Y.; Ko, H.J.; Ong, H.; Barrett, T.; Kim, J.K.; Davis, R.J. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol. Cell Biol. 2010, 30, 106–115. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Muscle as a secretory organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Silvis, C.; Deshpande, N.; Nystrom, G.; Frost, R.A. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock 2003, 19, 538–546. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Reyna, S.M.; Ghosh, S.; Tantiwong, P.; Meka, C.S.; Eagan, P.; Jenkinson, C.P.; Cersosimo, E.; Defronzo, R.A.; Coletta, D.K.; Sriwijitkamol, A.; et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 2008, 57, 2595–2602. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; McMillan, R.P.; Fausnacht, D.W.; Kavanaugh, J.W.; Harvey, M.M.; Stevens, J.R.; Wu, Y.; Mynatt, R.L.; Hulver, M.W. Muscle-specific Deletion of Toll-like Receptor 4 Impairs Metabolic Adaptation to Wheel Running in Mice. Med. Sci. Sports Exerc. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tancredi, R.G.; Dagenais, G.R.; Zierler, K.L. Free fatty acid metabolism in the forearm at rest: Muscle uptake and adipose tissue release of free fatty acids. Johns Hopkins Med. J. 1976, 138, 167–179. [Google Scholar] [PubMed]
- Kim, S.J.; Choi, Y.; Jun, H.S.; Kim, B.M.; Na, H.K.; Surh, Y.J.; Park, T. High-fat diet stimulates IL-1 type I receptor-mediated inflammatory signaling in the skeletal muscle of mice. Mol. Nutr. Food Res. 2010, 54, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, A.M.; Lenighan, Y.M.; O’Reilly, M.E.; McGillicuddy, F.C.; Roche, H.M. Nutritional modulation of metabolic inflammation. Biochem. Soc. Trans. 2017, 45, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Dixit, V.D. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 2010, 40, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef]
- Song, N.; Li, T. Regulation of NLRP3 Inflammasome by Phosphorylation. Front. Immunol. 2018, 9, 2305. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- McMurray, F.; Patten, D.A.; Harper, M.E. Reactive Oxygen Species and Oxidative Stress in Obesity-Recent Findings and Empirical Approaches. Obesity 2016, 24, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Campos, C.; Díaz-Vegas, A.; Galgani, J.E.; Juretic, N.; Osorio-Fuentealba, C.; Bucarey, J.L.; Tapia, G.; Valenzuela, R.; Contreras-Ferrat, A.; et al. Insulin-dependent H2O2 production is higher in muscle fibers of mice fed with a high-fat diet. Int. J. Mol. Sci. 2013, 14, 15740–15754. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox. Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, M.; Zhao, S.; Lu, Q.; Ni, L.; Zou, C.; Lu, L.; Xu, X.; Guan, H.; Zheng, Z.; et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflamm. Res. 2017, 66, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, Y.R.; Tang, T.T.; Pan, M.M.; Xu, S.C.; Ma, K.L.; Lv, L.L.; Liu, H.; Liu, B.C. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int. J. Biochem. Cell Biol. 2018, 98, 43–53. [Google Scholar] [CrossRef]
- Elshaer, S.L.; Mohamed, I.N.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.L.; Shanab, A.Y.; Lorys, R.; El-Remessy, A.B. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47. [Google Scholar] [CrossRef]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Schilling, W.P.; Wasylyna, T.; Dubyak, G.R.; Humphreys, B.D.; Sinkins, W.G. Maitotoxin and P2Z/P2X(7) purinergic receptor stimulation activate a common cytolytic pore. Am. J. Physiol. 1999, 277, C766–C776. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.B.; Rozovskaja, I.A.; Skulachev, V.P. Acidification of the interior of Ehrlich ascites tumor cells by nigericin inhibits DNA synthesis. FEBS Lett. 1987, 220, 288–290. [Google Scholar] [CrossRef]
- Pressman, B.C. Biological applications of ionophores. Annu. Rev. Biochem. 1976, 45, 501–530. [Google Scholar] [CrossRef]
- Adeva, M.M.; Souto, G. Diet-induced metabolic acidosis. Clin. Nutr. 2011, 30, 416–421. [Google Scholar] [CrossRef]
- Salameh, A.I.; Ruffin, V.A.; Boron, W.F. Effects of metabolic acidosis on intracellular pH responses in multiple cell types. Am. J. Physiol. Regul Integr. Comp. Physiol. 2014, 307, R1413–R1427. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Muramatsu, I.; Yasumoto, T. Na(+)-permeable channels induced by maitotoxin in guinea-pig single ventricular cells. Eur. J. Pharm. 1996, 297, 293–298. [Google Scholar] [CrossRef]
- Murata, M.; Gusovsky, F.; Yasumoto, T.; Daly, J.W. Selective stimulation of Ca2+ flux in cells by maitotoxin. Eur. J. Pharm. 1992, 227, 43–49. [Google Scholar] [CrossRef]
- Ohizumi, Y.; Kajiwara, A.; Yasumoto, T. Excitatory effect of the most potent marine toxin, maitotoxin, on the guinea-pig vas deferens. J. Pharm. Exp. Ther. 1983, 227, 199–204. [Google Scholar]
- Holmes, M.J.; Lewis, R.J. Purification and characterisation of large and small maitotoxins from cultured Gambierdiscus toxicus. Nat. Toxins 1994, 2, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Flores, P.L.; Rodríguez, E.; Zapata, E.; Carbó, R.; Farías, J.M.; Martínez, M. Maitotoxin Is a Potential Selective Activator of the Endogenous Transient Receptor Potential Canonical Type 1 Channel in Xenopus laevis Oocytes. Mar. Drugs 2017, 15, 198. [Google Scholar] [CrossRef] [PubMed]
- Skopin, A.; Shalygin, A.; Vigont, V.; Zimina, O.; Glushankova, L.; Mozhayeva, G.N.; Kaznacheyeva, E. TRPC1 protein forms only one type of native store-operated channels in HEK293 cells. Biochimie 2013, 95, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jaque-Fernandez, F.; Beaulant, A.; Berthier, C.; Monteiro, L.; Allard, B.; Casas, M.; Rieusset, J.; Jacquemond, V. Preserved Ca. Diabetologia 2020, 63, 2471–2481. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef]
- Sejersted, O.M.; Sjøgaard, G. Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol. Rev. 2000, 80, 1411–1481. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, L.; Canato, M.; Protasi, F.; Stienen, G.J.M.; Reggiani, C. A 3D diffusional-compartmental model of the calcium dynamics in cytosol, sarcoplasmic reticulum and mitochondria of murine skeletal muscle fibers. PLoS ONE 2018, 13, e0201050. [Google Scholar] [CrossRef]
- McBride, M.J.; Foley, K.P.; D’Souza, D.M.; Li, Y.E.; Lau, T.C.; Hawke, T.J.; Schertzer, J.D. The NLRP3 inflammasome contributes to sarcopenia and lower muscle glycolytic potential in old mice. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E222–E232. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Kny, M.; Riediger, F.; Busch, K.; Schmidt, S.; Luft, F.C.; Slevogt, H.; Fielitz, J. Deletion of Nlrp3 protects from inflammation-induced skeletal muscle atrophy. Intensive Care Med. Exp. 2017, 5, 3. [Google Scholar] [CrossRef]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Ruegg, U.T.; Takeda, S. ATP-Induced Increase in Intracellular Calcium Levels and Subsequent Activation of mTOR as Regulators of Skeletal Muscle Hypertrophy. Int. J. Mol. Sci. 2018, 19, 2804. [Google Scholar] [CrossRef]
- Coutinho-Silva, R.; Persechini, P.M. P2Z purinoceptor-associated pores induced by extracellular ATP in macrophages and J774 cells. Am. J. Physiol. 1997, 273, C1793–C1800. [Google Scholar] [CrossRef]
- Enjyoji, K.; Kotani, K.; Thukral, C.; Blumel, B.; Sun, X.; Wu, Y.; Imai, M.; Friedman, D.; Csizmadia, E.; Bleibel, W.; et al. Deletion of cd39/entpd1 results in hepatic insulin resistance. Diabetes 2008, 57, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jin, T. Extracellular high dosages of adenosine triphosphate induce inflammatory response and insulin resistance in rat adipocytes. Biochem. Biophys. Res. Commun. 2010, 402, 455–460. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Kobayashi, M.; Tagawa, R.; Nakagawa, Y.; Shimano, H.; Higami, Y. Association between Lysosomal Dysfunction and Obesity-Related Pathology: A Key Knowledge to Prevent Metabolic Syndrome. Int. J. Mol. Sci. 2019, 20, 3688. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Kusunoki, C.; Kondo, M.; Yasuda, M.; Kume, S.; Morino, K.; Sekine, O.; Ugi, S.; Uzu, T.; Nishio, Y.; et al. Autophagy regulates inflammation in adipocytes. Biochem. Biophys. Res. Commun. 2012, 417, 352–357. [Google Scholar] [CrossRef]
- Inami, Y.; Yamashina, S.; Izumi, K.; Ueno, T.; Tanida, I.; Ikejima, K.; Watanabe, S. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem. Biophys. Res. Commun. 2011, 412, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxid. Med. Cell Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef]
- Huang, X.; Vaag, A.; Carlsson, E.; Hansson, M.; Ahrén, B.; Groop, L. Impaired cathepsin L gene expression in skeletal muscle is associated with type 2 diabetes. Diabetes 2003, 52, 2411–2418. [Google Scholar] [CrossRef][Green Version]
- Chang, Y.C.; Liu, H.W.; Chen, Y.T.; Chen, Y.A.; Chen, Y.J.; Chang, S.J. Resveratrol protects muscle cells against palmitate-induced cellular senescence and insulin resistance through ameliorating autophagic flux. J. Food Drug Anal. 2018, 26, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: Roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Cell Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, M.A.; Saavedra, V.; Mangan, M.S.J.; Penno, A.; Thiele, C.; Latz, E.; Kuerschner, L. 1-Deoxysphingolipids cause autophagosome and lysosome accumulation and trigger NLRP3 inflammasome activation. Autophagy 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.T.; Koka, S.; Hussain, T.; Li, X. Simvastatin improves lysosome function via enhancing lysosome biogenesis in endothelial cells. Front. Biosci. 2020, 25, 283–298. [Google Scholar]
- Tang, T.T.; Lv, L.L.; Pan, M.M.; Wen, Y.; Wang, B.; Li, Z.L.; Wu, M.; Wang, F.M.; Crowley, S.D.; Liu, B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef]
- Naour, N.; Rouault, C.; Fellahi, S.; Lavoie, M.E.; Poitou, C.; Keophiphath, M.; Eberlé, D.; Shoelson, S.; Rizkalla, S.; Bastard, J.P.; et al. Cathepsins in human obesity: Changes in energy balance predominantly affect cathepsin s in adipose tissue and in circulation. J. Clin. Endocrinol. Metab. 2010, 95, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Colhoun, L.M.; Bains, B.K.; Kilgour, J.D.; Burden, R.E.; Burrows, J.F.; Lavelle, E.C.; Gilmore, B.F.; Scott, C.J. Extracellular cathepsin S and intracellular caspase 1 activation are surrogate biomarkers of particulate-induced lysosomal disruption in macrophages. Part. Fibre Toxicol. 2016, 13, 19. [Google Scholar] [CrossRef]
- Lafarge, J.C.; Pini, M.; Pelloux, V.; Orasanu, G.; Hartmann, G.; Venteclef, N.; Sulpice, T.; Shi, G.P.; Clément, K.; Guerre-Millo, M. Cathepsin S inhibition lowers blood glucose levels in mice. Diabetologia 2014, 57, 1674–1683. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.D. Gasdermin D Flashes an Exit Signal for IL-1. Immunity 2018, 48, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e36. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Saeki, N.; Kuwahara, Y.; Sasaki, H.; Satoh, H.; Shiroishi, T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome. 2000, 11, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Mitchell, S.J.; Wahl, D.; Diaz, A.; Singh, A.; Seo, W.; Wang, M.; Ali, A.; Kaiser, T.; Price, N.L.; et al. Disulfiram Treatment Normalizes Body Weight in Obese Mice. Cell Metab. 2020, 32, 203–214.e204. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Collino, M.; Benetti, E.; Rogazzo, M.; Mastrocola, R.; Yaqoob, M.M.; Aragno, M.; Thiemermann, C.; Fantozzi, R. Reversal of the deleterious effects of chronic dietary HFCS-55 intake by PPAR-δ agonism correlates with impaired NLRP3 inflammasome activation. Biochem. Pharm. 2013, 85, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Chiazza, F.; Couturier-Maillard, A.; Benetti, E.; Mastrocola, R.; Nigro, D.; Cutrin, J.C.; Serpe, L.; Aragno, M.; Fantozzi, R.; Ryffel, B.; et al. Targeting the NLRP3 Inflammasome to Reduce Diet-Induced Metabolic Abnormalities in Mice. Mol. Med. 2016, 21, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Kien, C.L.; Bunn, J.Y.; Fukagawa, N.K.; Anathy, V.; Matthews, D.E.; Crain, K.I.; Ebenstein, D.B.; Tarleton, E.K.; Pratley, R.E.; Poynter, M.E. Lipidomic evidence that lowering the typical dietary palmitate to oleate ratio in humans decreases the leukocyte production of proinflammatory cytokines and muscle expression of redox-sensitive genes. J. Nutr. Biochem. 2015, 26, 1599–1606. [Google Scholar] [CrossRef]
- Hull, C.; Dekeryte, R.; Buchanan, H.; Kamli-Salino, S.; Robertson, A.; Delibegovic, M.; Platt, B. NLRP3 inflammasome inhibition with MCC950 improves insulin sensitivity and inflammation in a mouse model of frontotemporal dementia. Neuropharmacology 2020, 180, 108305. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Wolff, S.M. The role of interleukin-1 in disease. N. Engl. J. Med. 1993, 328, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Esposito, K.; Marfella, R.; Giugliano, D. Plasma interleukin-18 concentrations are elevated in type 2 diabetes. Diabetes Care 2004, 27, 272. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulangé, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, H.; Haugen, F.; Zadelaar, S.; Kleemann, R.; Kooistra, T.; Drevon, C.A.; Blomhoff, R. Diet-induced obesity increases NF-kappaB signaling in reporter mice. Genes Nutr. 2009, 4, 215–222. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Krogh-Madsen, R.; Plomgaard, P.; Møller, K.; Mittendorfer, B.; Pedersen, B.K. Influence of TNF-alpha and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E108–E114. [Google Scholar] [CrossRef]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; Vasanthakumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Ekberg, K.; Landau, B.R. Lipid metabolism during fasting. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E789–E793. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef]
- Daemen, S.; Gemmink, A.; Brouwers, B.; Meex, R.C.R.; Huntjens, P.R.; Schaart, G.; Moonen-Kornips, E.; Jörgensen, J.; Hoeks, J.; Schrauwen, P.; et al. Distinct lipid droplet characteristics and distribution unmask the apparent contradiction of the athlete’s paradox. Mol. Metab. 2018, 17, 71–81. [Google Scholar] [CrossRef]
- van Loon, L.J. Use of intramuscular triacylglycerol as a substrate source during exercise in humans. J. Appl. Physiol. 2004, 97, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Dunville, K.; Li, J.; Cheng, J.X.; Conley, T.B.; Couture, C.S.; Campbell, W.W. Intermuscular Adipose Tissue Content and Intramyocellular Lipid Fatty Acid Saturation Are Associated with Glucose Homeostasis in Middle-Aged and Older Adults. Endocrinol. Metab. 2017, 32, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; He, J.; Watkins, S.; Kelley, D.E. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 2001, 86, 5755–5761. [Google Scholar] [CrossRef] [PubMed]
- Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Toledo, F.G.; Sauers, S.E.; Goodpaster, B.H. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E882–E888. [Google Scholar] [CrossRef]
- Samjoo, I.A.; Safdar, A.; Hamadeh, M.J.; Glover, A.W.; Mocellin, N.J.; Santana, J.; Little, J.P.; Steinberg, G.R.; Raha, S.; Tarnopolsky, M.A. Markers of skeletal muscle mitochondrial function and lipid accumulation are moderately associated with the homeostasis model assessment index of insulin resistance in obese men. PLoS ONE 2013, 8, e66322. [Google Scholar] [CrossRef]
- Nielsen, J.; Mogensen, M.; Vind, B.F.; Sahlin, K.; Højlund, K.; Schrøder, H.D.; Ortenblad, N. Increased subsarcolemmal lipids in type 2 diabetes: Effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E706–E713. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Goodpaster, B.H. Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity. Diabetes 2020, 69, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell-Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R.; et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef]
- Strle, K.; Broussard, S.R.; McCusker, R.H.; Shen, W.H.; Johnson, R.W.; Freund, G.G.; Dantzer, R.; Kelley, K.W. Proinflammatory cytokine impairment of insulin-like growth factor I-induced protein synthesis in skeletal muscle myoblasts requires ceramide. Endocrinology 2004, 145, 4592–4602. [Google Scholar] [CrossRef] [PubMed]
- Guillet, C.; Masgrau, A.; Walrand, S.; Boirie, Y. Impaired protein metabolism: Interlinks between obesity, insulin resistance and inflammation. Obes. Rev. 2012, 13 (Suppl. S2), 51–57. [Google Scholar] [CrossRef] [PubMed]
- Beals, J.W.; Burd, N.A.; Moore, D.R.; van Vliet, S. Obesity Alters the Muscle Protein Synthetic Response to Nutrition and Exercise. Front. Nutr. 2019, 6, 87. [Google Scholar] [CrossRef]
- Tardif, N.; Salles, J.; Guillet, C.; Tordjman, J.; Reggio, S.; Landrier, J.F.; Giraudet, C.; Patrac, V.; Bertrand-Michel, J.; Migne, C.; et al. Muscle ectopic fat deposition contributes to anabolic resistance in obese sarcopenic old rats through eIF2α activation. Aging Cell 2014, 13, 1001–1011. [Google Scholar] [CrossRef]
- Cho, K.A.; Kang, P.B. PLIN2 inhibits insulin-induced glucose uptake in myoblasts through the activation of the NLRP3 inflammasome. Int. J. Mol. Med. 2015, 36, 839–844. [Google Scholar] [CrossRef]
- Wu, K.K.; Cheung, S.W.; Cheng, K.K. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 4148. [Google Scholar] [CrossRef]
- Patsouris, D.; Cao, J.J.; Vial, G.; Bravard, A.; Lefai, E.; Durand, A.; Durand, C.; Chauvin, M.A.; Laugerette, F.; Debard, C.; et al. Insulin resistance is associated with MCP1-mediated macrophage accumulation in skeletal muscle in mice and humans. PLoS ONE 2014, 9, e110653. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Shi, H.; Chang, S.; Gao, Y.; Li, X.; Lv, C.; Yang, H.; Xiang, H.; Yang, J.; Xu, L.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int. Immunopharm. 2019, 74, 105575. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, X.X.; Xu, M. Inhibition of vascular neointima hyperplasia by FGF21 associated with FGFR1/Syk/NLRP3 inflammasome pathway in diabetic mice. Atherosclerosis 2019, 289, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Meng, X.; Ye, T.; Xie, W.; Sun, G.; Sun, X. Inhibiting the NLRP3 Inflammasome Activation with MCC950 Ameliorates Diabetic Encephalopathy in db/db Mice. Molecules 2018, 23, 522. [Google Scholar] [CrossRef] [PubMed]
- Trotta, M.C.; Maisto, R.; Guida, F.; Boccella, S.; Luongo, L.; Balta, C.; D’Amico, G.; Herman, H.; Hermenean, A.; Bucolo, C.; et al. The activation of retinal HCA2 receptors by systemic beta-hydroxybutyrate inhibits diabetic retinal damage through reduction of endoplasmic reticulum stress and the NLRP3 inflammasome. PLoS ONE 2019, 14, e0211005. [Google Scholar] [CrossRef]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 280. [Google Scholar] [CrossRef]
- Miyauchi, T.; Uchida, Y.; Kadono, K.; Hirao, H.; Kawasoe, J.; Watanabe, T.; Ueda, S.; Okajima, H.; Terajima, H.; Uemoto, S. Up-regulation of FOXO1 and reduced inflammation by β-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc. Natl. Acad. Sci. USA 2019, 116, 13533–13542. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, H.H.; Rittig, N.; Johannsen, M.; Møller, A.B.; Jørgensen, J.O.; Jessen, N.; Møller, N. Effects of 3-hydroxybutyrate and free fatty acids on muscle protein kinetics and signaling during LPS-induced inflammation in humans: Anticatabolic impact of ketone bodies. Am. J. Clin. Nutr. 2018, 108, 857–867. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorquera, G.; Russell, J.; Monsalves-Álvarez, M.; Cruz, G.; Valladares-Ide, D.; Basualto-Alarcón, C.; Barrientos, G.; Estrada, M.; Llanos, P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 3254. https://doi.org/10.3390/ijms22063254
Jorquera G, Russell J, Monsalves-Álvarez M, Cruz G, Valladares-Ide D, Basualto-Alarcón C, Barrientos G, Estrada M, Llanos P. NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. International Journal of Molecular Sciences. 2021; 22(6):3254. https://doi.org/10.3390/ijms22063254
Chicago/Turabian StyleJorquera, Gonzalo, Javier Russell, Matías Monsalves-Álvarez, Gonzalo Cruz, Denisse Valladares-Ide, Carla Basualto-Alarcón, Genaro Barrientos, Manuel Estrada, and Paola Llanos. 2021. "NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle" International Journal of Molecular Sciences 22, no. 6: 3254. https://doi.org/10.3390/ijms22063254
APA StyleJorquera, G., Russell, J., Monsalves-Álvarez, M., Cruz, G., Valladares-Ide, D., Basualto-Alarcón, C., Barrientos, G., Estrada, M., & Llanos, P. (2021). NLRP3 Inflammasome: Potential Role in Obesity Related Low-Grade Inflammation and Insulin Resistance in Skeletal Muscle. International Journal of Molecular Sciences, 22(6), 3254. https://doi.org/10.3390/ijms22063254