
Iron Availability in Tissue Microenvironment: The Key Role of Ferroportin


{kind=link}
Abstract
1. Introduction
2. Control of Iron Homeostasis
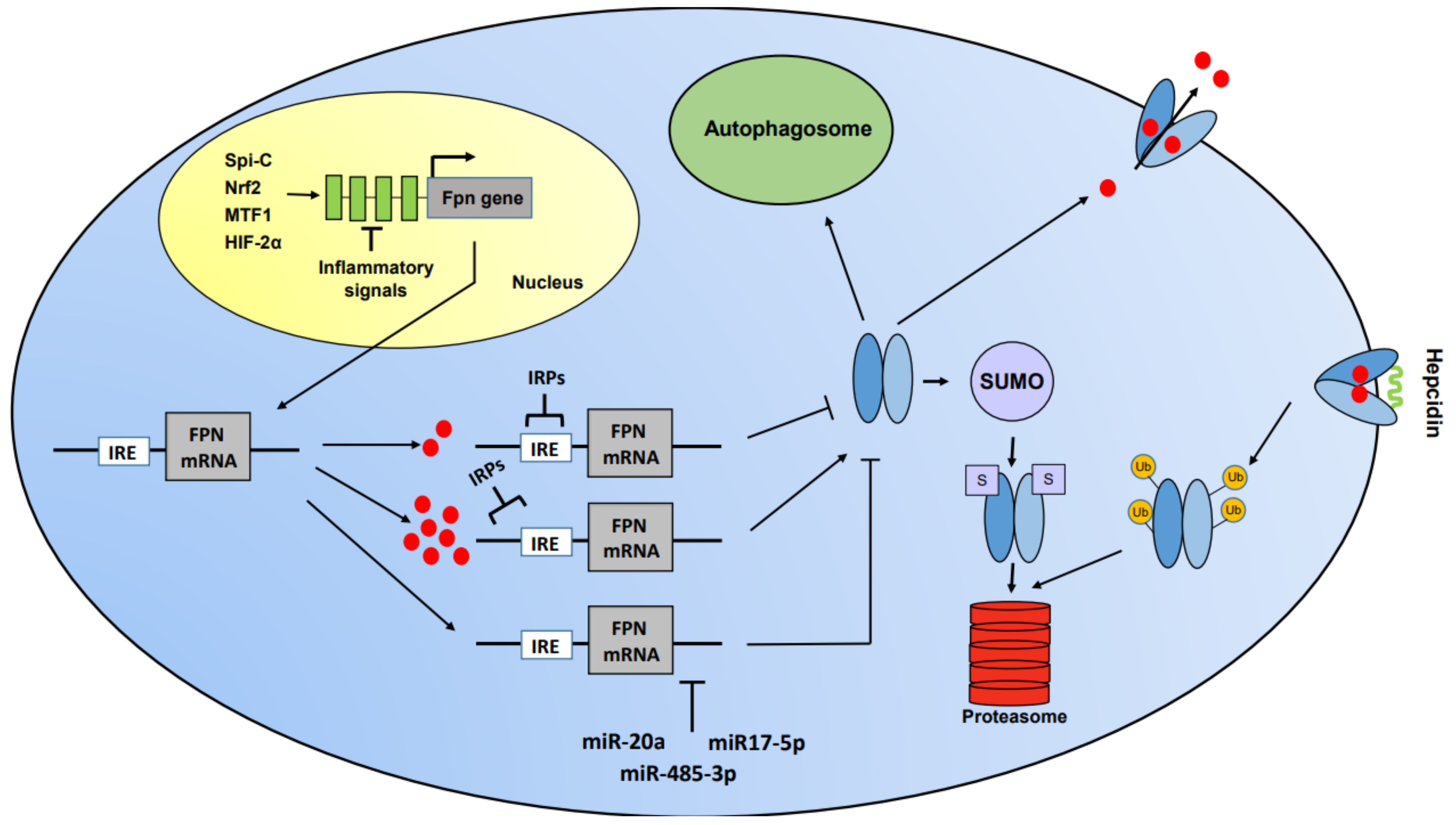
3. Ferroportin
3.1. Lessons from Clinical Studies and Mouse Models
4. Role of Ferroportin-Mediated Modulation of Iron Availability in Tissue Microenvironment
4.1. Heart
4.2. Intestine
4.3. Lung
4.4. Placenta-Fetus
4.5. Liver
4.6. Hair
5. Role of Ferroportin-Mediated Modulation of Local Iron Availability in Pathological Settings
5.1. Infection-Inflammation
5.2. Cancer
5.3. Wound Healing
5.4. Atherosclerosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COPD | Chronic obstructive pulmonary disease |
| CSC | Cancer stem cells |
| FD | Ferroportin disease |
| FPN | Ferroportin |
| HIF | Hypoxia-inducible factor |
| IRE | Iron regulatory element |
| IRP | Iron regulatory protein |
| MTF1 | Metal-regulatory transcription factor 1 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| ROS | Reactive oxygen species |
| SUMO | Small ubiquitin-like modifier |
| TAM | Tumor-associated macrophage |
| TLR | Toll-like receptor |
| TfR1 | Transferrin receptor |
References
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Golonka, R.; Yeoh, B.S.; Vijay-Kumar, M. The Iron Tug-of-War between Bacterial Siderophores and Innate Immunity. J. Innate Immun. 2019, 11, 249–262. [Google Scholar] [CrossRef]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxidative Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging. Elife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Timmers, P.R.H.J.; Wilson, J.F.; Joshi, P.K.; Deelen, J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Daghlas, I.; Gill, D. Genetically predicted iron status and life expectancy. Clin. Nutr. 2020. [Google Scholar] [CrossRef]
- Herlihy, J.H.; Long, T.A.; McDowell, J.M. Iron homeostasis and plant immune responses: Recent insights and translational implications. J. Biol. Chem. 2020, 295, 13444–13457. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. Macrophages: Central regulators of iron balance. Metallomics 2014, 6, 1336–1345. [Google Scholar] [CrossRef]
- Bories, G.F.P.; Yeudall, S.; Serbulea, V.; Fox, T.E.; Isakson, B.E.; Leitinger, N. Macrophage metabolic adaptation to heme detoxification involves CO-dependent activation of the pentose phosphate pathway. Blood 2020, 136, 1535–1548. [Google Scholar] [CrossRef]
- Pek, R.H.; Yuan, X.; Rietzschel, N.; Zhang, J.; Jackson, L.; Nishibori, E.; Ribeiro, A.; Simmons, W.; Jagadeesh, J.; Sugimoto, H.; et al. Hemozoin produced by mammals confers heme tolerance. Elife 2019, 8, e49503. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e574. [Google Scholar] [CrossRef]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Kato, H.; Hada, H.; Itoh-Nakadai, A.; Fujiwara, T.; Muto, A.; Inoguchi, Y.; Ichiyanagi, K.; Hojo, W.; Tomosugi, N.; et al. Iron-heme-Bach1 axis is involved in erythroblast adaptation to iron deficiency. Haematologica 2017, 102, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Rifkind, J.M.; Nagababu, E.; Ramasamy, S.; Ravi, L.B. Hemoglobin redox reactions and oxidative stress. Redox Rep. 2003, 8, 234–237. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural basis of ion transport and inhibition in ferroportin. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, V.; Bonaccorsi di Patti, M.C.; Iacovelli, F.; Pasquadibisceglie, A.; Falconi, M.; Musci, G.; Polticelli, F. Dynamical Behavior of the Human Ferroportin Homologue from. Int. J. Mol. Sci. 2020, 21, 6785. [Google Scholar] [CrossRef]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef]
- Yang, X.; Park, S.H.; Chang, H.C.; Shapiro, J.S.; Vassilopoulos, A.; Sawicki, K.T.; Chen, C.; Shang, M.; Burridge, P.W.; Epting, C.L.; et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J. Clin. Invest. 2017, 127, 1505–1516. [Google Scholar] [CrossRef]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2009, 457, 318–321. [Google Scholar] [CrossRef]
- Chen, P.H.; Wu, J.; Ding, C.C.; Lin, C.C.; Pan, S.; Bossa, N.; Xu, Y.; Yang, W.H.; Mathey-Prevot, B.; Chi, J.T. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020, 27, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Troadec, M.B.; Ward, D.M.; Lo, E.; Kaplan, J.; De Domenico, I. Induction of FPN1 transcription by MTF-1 reveals a role for ferroportin in transition metal efflux. Blood 2010, 116, 4657–4664. [Google Scholar] [CrossRef]
- Yang, F.; Liu, X.B.; Quinones, M.; Melby, P.C.; Ghio, A.; Haile, D.J. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J. Biol. Chem. 2002, 277, 39786–39791. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.; Devalaraja, S.; Li, M.; To, T.K.J.; Folkert, I.W.; Mitchell-Velasquez, E.; Dang, M.T.; Young, P.; Wilbur, C.J.; Silverman, M.A.; et al. Counter Regulation of Spic by NF-κB and STAT Signaling Controls Inflammation and Iron Metabolism in Macrophages. Cell Rep. 2020, 31, 107825. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Gammella, E.; Buratti, P.; Doni, A.; Anselmo, A.; Locati, M.; Cairo, G. Macrophage ferroportin is essential for stromal cell proliferation in wound healing. Haematologica 2019, 104, 47–58. [Google Scholar] [CrossRef]
- Helgudottir, S.S.; Routhe, L.J.; Burkhart, A.; Jønsson, K.; Pedersen, I.S.; Lichota, J.; Moos, T. Epigenetic Regulation of Ferroportin in Primary Cultures of the Rat Blood-Brain Barrier. Mol. Neurobiol. 2020, 57, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Sangokoya, C.; Doss, J.F.; Chi, J.T. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013, 9, e1003408. [Google Scholar] [CrossRef]
- Babu, K.R.; Muckenthaler, M.U. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J. Mol. Med. 2016, 94, 347–359. [Google Scholar] [CrossRef]
- Link, C.; Knopf, J.D.; Marques, O.; Lemberg, M.K.; Muckenthaler, M.U. The role of cellular iron deficiency in controlling iron export. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129829. [Google Scholar] [CrossRef] [PubMed]
- Bayele, H.K.; Srai, S.K.S. A disease-causing mutation K240E disrupts ferroportin trafficking by SUMO (ferroportin SUMOylation). Biochem. Biophys. Rep. 2021, 25, 100873. [Google Scholar]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X.; et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Jung, G.; Gabayan, V.; Valore, E.; Ruchala, P.; Gulig, P.A.; Ganz, T.; Nemeth, E.; Bulut, Y. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 2015, 17, 47–57. [Google Scholar] [CrossRef]
- Guida, C.; Altamura, S.; Klein, F.A.; Galy, B.; Boutros, M.; Ulmer, A.J.; Hentze, M.W.; Muckenthaler, M.U. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 2015, 125, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Enculescu, M.; Metzendorf, C.; Sparla, R.; Hahnel, M.; Bode, J.; Muckenthaler, M.U.; Legewie, S. Modelling Systemic Iron Regulation during Dietary Iron Overload and Acute Inflammation: Role of Hepcidin-Independent Mechanisms. PLoS Comput. Biol. 2017, 13, e1005322. [Google Scholar] [CrossRef]
- Vlasveld, L.T.; Janssen, R.; Bardou-Jacquet, E.; Venselaar, H.; Hamdi-Roze, H.; Drakesmith, H.; Swinkels, D.W. Twenty Years of Ferroportin Disease: A Review or An Update of Published Clinical, Biochemical, Molecular, and Functional Features. Pharmaceuticals 2019, 12, 132. [Google Scholar] [CrossRef]
- Pietrangelo, A. Ferroportin disease: Pathogenesis, diagnosis and treatment. Haematologica 2017, 102, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Sabelli, M.; Montosi, G.; Garuti, C.; Caleffi, A.; Oliveto, S.; Biffo, S.; Pietrangelo, A. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 2017, 65, 1512–1525. [Google Scholar] [CrossRef]
- Tangudu, N.K.; Yilmaz, D.; Wörle, K.; Gruber, A.; Colucci, S.; Leopold, K.; Muckenthaler, M.U.; Vujic Spasic, M. Macrophage-HFE controls iron metabolism and immune responses in aged mice. Haematologica 2021, 106, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Sweetland, E.; Schimanski, L.; Edwards, J.; Cowley, D.; Ashraf, M.; Bastin, J.; Townsend, A.R. The hemochromatosis protein HFE inhibits iron export from macrophages. Proc. Natl. Acad. Sci. USA 2002, 99, 15602–15607. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Montosi, G.; Castrusini, E.; Conte, D.; Pietrangelo, A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood 1997, 89, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Winn, N.C.; Volk, K.M.; Hasty, A.H. Regulation of tissue iron homeostasis: The macrophage “ferrostat”. JCI Insight 2020, 5, e132964. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; An, P.; Min, J.; Wang, F. Cardiomyocyte-specific deletion of ferroportin using MCK-Cre has no apparent effect on cardiac iron homeostasis. Int. J. Cardiol. 2015, 201, 90–92. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S. Mechanisms of cardiac iron homeostasis and their importance to heart function. Free Radic. Biol. Med. 2019, 133, 234–237. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Converso-Baran, K.; Michele, D.E.; Shah, Y.M. A genetic mouse model of severe iron deficiency anemia reveals tissue-specific transcriptional stress responses and cardiac remodeling. J. Biol. Chem. 2019, 294, 14991–15002. [Google Scholar] [CrossRef] [PubMed]
- Bessman, N.J.; Mathieu, J.R.R.; Renassia, C.; Zhou, L.; Fung, T.C.; Fernandez, K.C.; Austin, C.; Moeller, J.B.; Zumerle, S.; Louis, S.; et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 2020, 368, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J. Clin. Invest. 2019, 129, 336–348. [Google Scholar] [CrossRef]
- Ghio, A.J. Disruption of iron homeostasis and lung disease. Biochim. Biophys. Acta 2009, 1790, 731–739. [Google Scholar] [CrossRef]
- Neves, J.; Leitz, D.; Kraut, S.; Brandenberger, C.; Agrawal, R.; Weissmann, N.; Mühlfeld, C.; Mall, M.A.; Altamura, S.; Muckenthaler, M.U. Disruption of the Hepcidin/Ferroportin Regulatory System Causes Pulmonary Iron Overload and Restrictive Lung Disease. EBioMedicine 2017, 20, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.; Baker, J.R.; Di Giandomenico, S.; Kermani, P.; Parker, J.; Kim, K.; Yang, J.; Barnes, P.J.; Vaulont, S.; Scandura, J.M.; et al. Hepcidin Is Essential for Alveolar Macrophage Function and Is Disrupted by Smoke in a Murine Chronic Obstructive Pulmonary Disease Model. J. Immunol. 2020, 205, 2489–2498. [Google Scholar] [CrossRef]
- Yang, F.; Haile, D.J.; Wang, X.; Dailey, L.A.; Stonehuerner, J.G.; Ghio, A.J. Apical location of ferroportin 1 in airway epithelia and its role in iron detoxification in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L14–L23. [Google Scholar] [CrossRef]
- Hazlett, H.F.; Hampton, T.H.; Aridgides, D.S.; Armstrong, D.A.; Dessaint, J.A.; Mellinger, D.L.; Nymon, A.B.; Ashare, A. Altered iron metabolism in cystic fibrosis macrophages: The impact of CFTR modulators and implications for Pseudomonas aeruginosa survival. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Leibold, E.A.; Harris, Z.L.; Wobken, J.D.; Clarke, S.; Zumbrennen, K.B.; Eisenstein, R.S.; Georgieff, M.K. Influence of gestational age and fetal iron status on IRP activity and iron transporter protein expression in third-trimester human placenta. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R894–R901. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Fisher, A.L.; Chua, K.J.; Ruchala, P.; Ganz, T.; Nemeth, E. Maternal hepcidin determines embryo iron homeostasis in mice. Blood 2020, 136, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Sangkhae, V.; Fisher, A.L.; Wong, S.; Koenig, M.D.; Tussing-Humphreys, L.; Chu, A.; Lelić, M.; Ganz, T.; Nemeth, E. Effects of maternal iron status on placental and fetal iron homeostasis. J. Clin. Invest. 2020, 130, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Kämmerer, L.; Mohammad, G.; Wolna, M.; Robbins, P.A.; Lakhal-Littleton, S. Fetal liver hepcidin secures iron stores in utero. Blood 2020, 136, 1549–1557. [Google Scholar] [CrossRef]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef]
- Han, C.Y.; Koo, J.H.; Kim, S.H.; Gardenghi, S.; Rivella, S.; Strnad, P.; Hwang, S.J.; Kim, S.G. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Trost, L.B.; Bergfeld, W.F.; Calogeras, E. The diagnosis and treatment of iron deficiency and its potential relationship to hair loss. J. Am. Acad. Derm. 2006, 54, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Bennoun, M.; Porteu, A.; Mativet, S.; Beaumont, C.; Grandchamp, B.; Sirito, M.; Sawadogo, M.; Kahn, A.; Vaulont, S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. USA 2002, 99, 4596–4601. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; She, E.; Gelbart, T.; Truksa, J.; Lee, P.; Xia, Y.; Khovananth, K.; Mudd, S.; Mann, N.; Moresco, E.M.; et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008, 320, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; de Lara, F.M.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.; Quinn, F.; Giri, P.K. Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. 2018, 2, 1089–1100. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Gammella, E.; Invernizzi, P.; Cairo, G. Iron levels in polarized macrophages: Regulation of immunity and autoimmunity. Autoimmun. Rev. 2012, 11, 883–889. [Google Scholar] [CrossRef]
- Recalcati, S.; Gammella, E.; Cairo, G. Ironing out Macrophage Immunometabolism. Pharmaceuticals 2019, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Chen, T.D.; Buang, N.; Olona, A.; Ko, J.H.; Prendecki, M.; Costa, A.S.H.; Nikitopoulou, E.; Tronci, L.; Pusey, C.D.; et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep. 2019, 28, 498–511. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Nairz, M.; Haschka, D.; Demetz, E.; Weiss, G. Iron at the interface of immunity and infection. Front. Pharm. 2014, 5, 152. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Farrell, T.J.; Trothen, S.M.; Dikeakos, J.D.; Heinrichs, D.E. Rapid removal of phagosomal ferroportin in macrophages contributes to nutritional immunity. Blood Adv. 2021, 5, 459–474. [Google Scholar] [CrossRef]
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115–130. [Google Scholar] [CrossRef]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Remsik, J.; Kiseliovas, V.; Derderian, C.; Sener, U.; Alghader, M.; Saadeh, F.; Nikishina, K.; Bale, T.; Iacobuzio-Donahue, C.; et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020, 369, 276–282. [Google Scholar]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Blanchette-Farra, N.; Kita, D.; Konstorum, A.; Tesfay, L.; Lemler, D.; Hegde, P.; Claffey, K.P.; Torti, F.M.; Torti, S.V. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018, 37, 4013–4032. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Gammella, E.; Cairo, G. Dysregulation of iron metabolism in cancer stem cells. Free Radic. Biol. Med. 2019, 133, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Corna, G.; Caserta, I.; Monno, A.; Apostoli, P.; Manfredi, A.A.; Camaschella, C.; Rovere-Querini, P. The Repair of Skeletal Muscle Requires Iron Recycling through Macrophage Ferroportin. J. Immunol. 2016, 197, 1914–1925. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 2011, 121, 985–997. [Google Scholar] [CrossRef]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef]
- Malhotra, R.; Wunderer, F.; Barnes, H.J.; Bagchi, A.; Buswell, M.D.; O’Rourke, C.D.; Slocum, C.L.; Ledsky, C.D.; Peneyra, K.M.; Sigurslid, H.; et al. Hepcidin Deficiency Protects Against Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Finn, A.V. The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis. Front. Pharm. 2014, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Bories, G.; Colin, S.; Vanhoutte, J.; Derudas, B.; Copin, C.; Fanchon, M.; Daoudi, M.; Belloy, L.; Haulon, S.; Zawadzki, C.; et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ. Res. 2013, 113, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Invest. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, M.; Liu, Y.; Li, H.; Shang, L.; Xu, T.; Chen, Z.; Wang, F.; Qiao, T.; Li, K. Iron accumulation in macrophages promotes the formation of foam cells and development of atherosclerosis. Cell Biosci. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; van Lier, J.J.; Roubert, B.; Haboubi, T.; Göhring, U.M.; Dürrenberger, F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am. J. Hematol. 2020, 95, 68–77. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gammella, E.; Correnti, M.; Cairo, G.; Recalcati, S. Iron Availability in Tissue Microenvironment: The Key Role of Ferroportin. Int. J. Mol. Sci. 2021, 22, 2986. https://doi.org/10.3390/ijms22062986
Gammella E, Correnti M, Cairo G, Recalcati S. Iron Availability in Tissue Microenvironment: The Key Role of Ferroportin. International Journal of Molecular Sciences. 2021; 22(6):2986. https://doi.org/10.3390/ijms22062986
Chicago/Turabian StyleGammella, Elena, Margherita Correnti, Gaetano Cairo, and Stefania Recalcati. 2021. "Iron Availability in Tissue Microenvironment: The Key Role of Ferroportin" International Journal of Molecular Sciences 22, no. 6: 2986. https://doi.org/10.3390/ijms22062986
APA StyleGammella, E., Correnti, M., Cairo, G., & Recalcati, S. (2021). Iron Availability in Tissue Microenvironment: The Key Role of Ferroportin. International Journal of Molecular Sciences, 22(6), 2986. https://doi.org/10.3390/ijms22062986