
Tryptophan Metabolism and Gut-Brain Homeostasis



Abstract
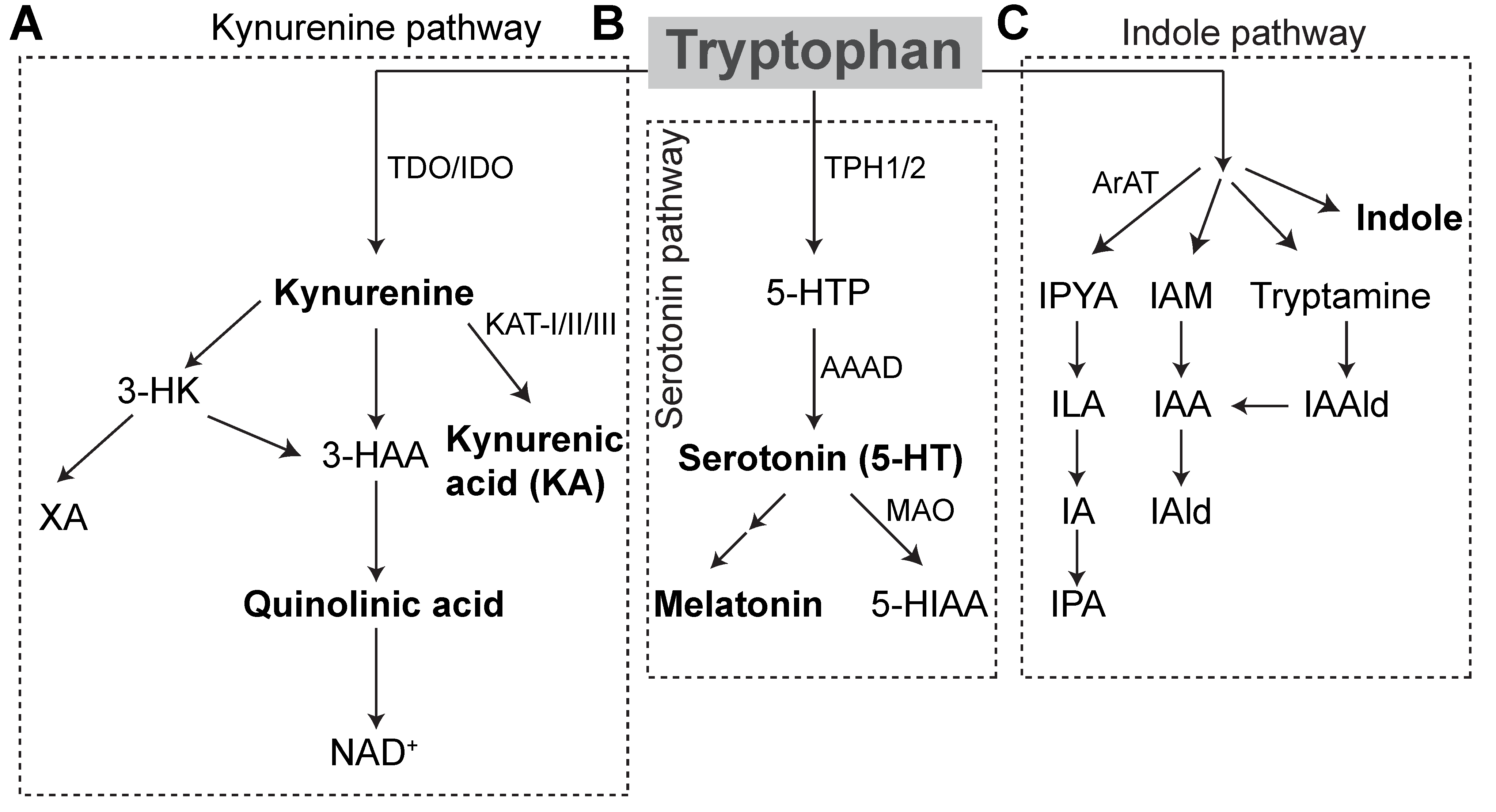
1. Tryptophan
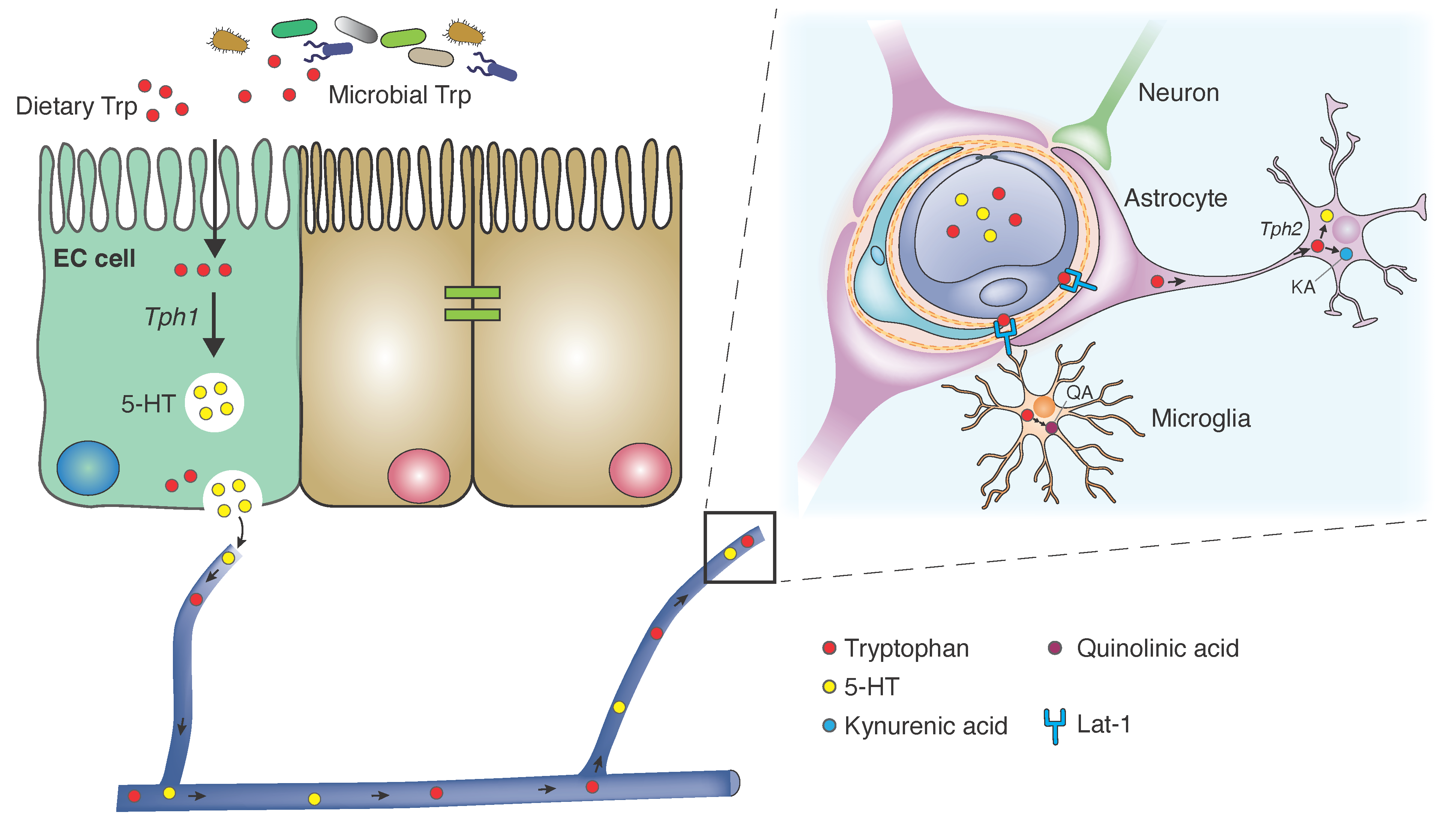
1.1. Dietary Intake and Absorption
1.2. Host Metabolic Pathways
1.2.1. Serotonin Pathway
1.2.2. Kynurenine Pathway
2. Tryptophan Metabolites
2.1. Serotonin
2.1.1. Homeostasis
2.1.2. Gut Signaling
2.1.3. Gut Motility
2.1.4. Vasoreactivity
2.1.5. Platelet Activation
2.1.6. Immune Cell Activity
2.1.7. Central Serotonin Activity

{kind=link}
{kind=link}
| Receptor Subtype | Primary Locations | Implicated Disorders |
|---|---|---|
| 5-HT1A | CNS (Median raphe nuclei; septal nuclei; hippocampus; neocortex) | Anxiety and depression |
| 5-HT1B | CNS (Ventral pallidum; substantia nigra), intracranial vasculature | Migraine |
| 5-HT1D | CNS (Ventral pallidum; substantia nigra), intracranial vasculature | Migraine |
| 5-HT1E | CNS (Olfactory bulb; hippocampus; neocortex; striatum) | ? |
| 5-HTF | CNS (Trigeminal ganglia; neocortex; hippocampus; astroglia), PNS (Dorsal root ganglia), Renal proximal tubule, coronary artery, pulmonary artery | Migraine |
| 5-HT2A | CNS (Neocortex); vascular smooth muscle | Psychosis, schizophrenia, hypertension |
| 5-HT2B | Cardiac Fibroblasts, stomach | Heart failure, anxiety, pulmonary hypertension |
| 5-HT2C (formerly 5-HT1C) | CNS (Choroid plexus) | Obesity, epilepsy, psychosis, mood disorders, anxiety |
| 5-HT3 | CNS (Substantia gelatinosa; area postrema) | Nausea and vomiting |
| 5-HT4 | CNS (Hippocampus; colliculi), GI tract | GI motility disorders |
| 5-HT5 | CNS (Hippocampus) | Sleep disorders |
| 5-HT6 | CNS (Striatum; neocortex; limbic system) | Cognitive disorders, obesity, seizures |
| 5-HT7 | CNS (Suprachiasmatic nucleus; hippocampus; thalamus) | Anxiety, sleep disorders, cognitive disorders |
2.2. Kynurenine
2.2.1. Central Activity of Kynurenine Metabolites
2.2.2. Kynurenic Acid
2.2.3. Quinolinic Acid
2.3. Gut Microbial Tryptophan Metabolism
2.3.1. Studying Tryptophan Metabolisms in Animal Models
2.3.2. Indole Pathway
2.3.3. Tryptophan Biosynthesis
3. Tryptophan Metabolites in Neurodevelopment, Neurologic and Psychiatric Disorders
3.1. Serotonin and Neurodevelopment
3.2. Serotonin and Cognition
3.3. Neurological Disorders
3.3.1. Alzheimer’s Disease
3.3.2. Parkinson’s Disease
3.3.3. Other Neurodegenerative Disorders
3.3.4. Multiple Sclerosis
3.3.5. Cerebrovascular Disease
3.3.6. Migraine
3.3.7. Traumatic Brain Injury
3.3.8. Minocycline in Brain Injury
3.4. Psychiatric Disorders
3.4.1. Anxiety and Depression
3.4.2. Schizophrenia
3.4.3. Autism Spectrum Disorders
3.5. Gastrointestinal Disorders
3.5.1. Irritable Bowel Syndrome
3.5.2. Inflammatory Bowel Disease
3.5.3. Age-Related Gastrointestinal Dysfunction
4. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Strasser, B.; Gostner, J.M.; Fuchs, D. Mood, food, and cognition: Role of tryptophan and serotonin. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 55–61. [Google Scholar] [CrossRef]
- Shabbir, F.; Patel, A.; Mattison, C.; Bose, S.; Krishnamohan, R.; Sweeney, E.; Sandhu, S.; Nel, W.; Rais, A.; Sandhu, R. Effect of diet on serotonergic neurotransmission in depression. Neurochem. Int. 2013, 62, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Ruiz-Canela, M.; Guasch-Ferré, M.; Zheng, Y.; Toledo, E.; Clish, C.B.; Salas-Salvadó, J.; Liang, L.; Wang, D.D.; Corella, D. Increases in plasma tryptophan are inversely associated with incident cardiovascular disease in the Prevencion con Dieta Mediterranea (PREDIMED) Study. J. Nutr. 2017, 147, 314–322. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.-I.; Farzi, A.; Zhu, W.-Y. Tryptophan metabolism: A link between the gut microbiota and brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, L.; Clarke, G.; Nolan, A.; Watkins, C.; Dinan, T.G.; Stanton, C.; Ross, R.P.; Ryan, C.A. Tryptophan metabolic profile in term and preterm breast milk: Implications for health. J. Nutr. Sci. 2018, 7, e13. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, D.; Troost, F.; Masclee, A. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Fierer, G. Transport of tryptophan into brain from the circulating, albumin-bound pool in rats and in rabbits. J. Neurochem. 1990, 54, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Ruddick, J.P.; Evans, A.K.; Nutt, D.J.; Lightman, S.L.; Rook, G.A.; Lowry, C.A. Tryptophan metabolism in the central nervous system: Medical implications. Expert Rev. Mol. Med. 2006, 8, 1. [Google Scholar] [CrossRef]
- Boadle-Biber, M.C. Regulation of serotonin synthesis. Prog. Biophys. Mol. Biol. 1993, 60, 1–15. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.; Dinan, T.; Cryan, J. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2, 3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy states. Int. J. Tryptophan Res. 2009, 2, S2097. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.J.F.R., III; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Gershon, M.; Dreyfus, C.; Pickel, V.; Joh, T.; Reis, D. Serotonergic neurons in the peripheral nervous system: Identification in gut by immunohistochemical localization of tryptophan hydroxylase. Proc. Natl. Acad. Sci. USA 1977, 74, 3086–3089. [Google Scholar] [CrossRef] [PubMed]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut-functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473. [Google Scholar] [CrossRef]
- Browning, K.N. Role of central vagal 5-HT3 receptors in gastrointestinal physiology and pathophysiology. Front. Neurosci. 2015, 9, 413. [Google Scholar] [CrossRef]
- Hesketh, P.J.; Gandara, D.R. Serotonin antagonists: A new class of antiemetic agents. JNCI J. Natl. Cancer Inst. 1991, 83, 613–620. [Google Scholar] [CrossRef]
- Coates, M.D.; Johnson, A.C.; Greenwood-van Meerveld, B.; Mawe, G.M. Effects of serotonin transporter inhibition on gastrointestinal motility and colonic sensitivity in the mouse. Neurogastroenterol. Motil. 2006, 18, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Heredia, D.J.; Gershon, M.D.; Koh, S.D.; Corrigan, R.D.; Okamoto, T.; Smith, T.K. Important role of mucosal serotonin in colonic propulsion and peristaltic reflexes: In vitro analyses in mice lacking tryptophan hydroxylase 1. J. Physiol. 2013, 591, 5939–5957. [Google Scholar] [CrossRef]
- Ellis, M.; Chambers, J.D.; Gwynne, R.M.; Bornstein, J.C. Serotonin and cholecystokinin mediate nutrient-induced segmentation in guinea pig small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G749–G761. [Google Scholar] [CrossRef] [PubMed]
- Tonini, M.; Pace, F. Drugs acting on serotonin receptors for the treatment of functional GI disorders. Dig. Dis. 2006, 24, 59–69. [Google Scholar] [CrossRef]
- Zhu, J.; Wu, X.; Owyang, C.; Li, Y. Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat. J. Physiol. 2001, 530, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Giordano, J.; Dyche, J. Differential analgesic actions of serotonin 5-HT3 receptor antagonists in the mouse. Neuropharmacology 1989, 28, 423–427. [Google Scholar] [CrossRef]
- Crowell, M.D. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br. J. Pharmacol. 2004, 141, 1285–1293. [Google Scholar] [CrossRef]
- Ma, L.; Yu, Z.; Xiao, S.; Thadani, U.; Robinson, C.P.; Patterson, E. Supersensitivity to serotonin-and histamine-induced arterial contraction following ovariectomy. Eur. J. Pharmacol. 1998, 359, 191–200. [Google Scholar] [CrossRef]
- Sobey, C.G.; Dusting, G.J.; Woodman, O.L. Enhanced vasoconstriction by serotonin in rabbit carotid arteries with atheroma-like lesions in vivo. Clin. Exp. Pharmacol. Physiol. 1991, 18, 367–370. [Google Scholar] [CrossRef]
- Bolte, A.C.; Van Geijn, H.P.; Dekker, G.A. Pathophysiology of preeclampsia and the role of serotonin. Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 95, 12–21. [Google Scholar] [CrossRef]
- Mohammad-Zadeh, L.; Moses, L.; Gwaltney-Brant, S. Serotonin: A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef]
- Cerrito, E.; Lazzaro, M.; Gaudio, E.; Arminio, P.; Aloisi, G. 5HT2-receptors and serotonin release: Their role in human platelet aggregation. Life Sci. 1993, 53, 209–215. [Google Scholar] [CrossRef]
- Herr, N.; Bode, C.; Duerschmied, D. The effects of serotonin in immune cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246250. [Google Scholar] [CrossRef]
- Idzko, M.; Panther, E.; Stratz, C.; Müller, T.; Bayer, H.; Zissel, G.; Dürk, T.; Sorichter, S.; Di Virgilio, F.; Geissler, M. The serotoninergic receptors of human dendritic cells: Identification and coupling to cytokine release. J. Immunol. 2004, 172, 6011–6019. [Google Scholar] [CrossRef]
- Hellstrand, K.; Czerkinsky, C.; Ricksten, A.; Jansson, B.; Asea, A.; Kylefjord, H.; Hermodsson, S. Role of serotonin in the regulation of interferon-γ production by human natural killer cells. J. Interferon Res. 1993, 13, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mössner, R.; Lesch, K.-P. Role of serotonin in the immune system and in neuroimmune interactions. Brain Behav. Immun. 1998, 12, 249–271. [Google Scholar] [CrossRef]
- Fiebich, B.; Akundi, R.; Lieb, K.; Candelario-Jalil, E.; Gmeiner, D.; Haus, U.; Müller, W.; Stratz, T.; Munoz, E. Antiinflammatory effects of 5-HT3 receptor antagonists in lipopolysaccharide-stimulated primary human monocytes. Scand. J. Rheumatol. 2004, 33, 28–32. [Google Scholar] [CrossRef]
- Frankiensztajn, L.M.; Elliott, E.; Koren, O. The microbiota and the hypothalamus-pituitary-adrenocortical (HPA) axis, implications for anxiety and stress disorders. Curr. Opin. Neurobiol. 2020, 62, 76–82. [Google Scholar] [CrossRef]
- Francis-Oliveira, J.; Shieh, I.; Higa, G.V.; Barbosa, M.; De Pasquale, R. Maternal separation induces changes in TREK-1 and 5HT1A expression in brain areas involved in the stress response in a sex-dependent way. Behav. Brain Res. 2020, 396, 112909. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, H.J.; Kim, J.G.; Ryu, V.; Kim, B.-T.; Kang, D.-W.; Jahng, J.W. Depressive behaviors and decreased expression of serotonin reuptake transporter in rats that experienced neonatal maternal separation. Neurosci. Res. 2007, 58, 32–39. [Google Scholar] [CrossRef]
- Lieb, M.W.; Weidner, M.; Arnold, M.R.; Loupy, K.M.; Nguyen, K.T.; Hassell, J.E., Jr.; K’Loni, S.S.; Kern, R.; Day, H.E.; Lesch, K.-P. Effects of maternal separation on serotonergic systems in the dorsal and median raphe nuclei of adult male Tph2-deficient mice. Behav. Brain Res. 2019, 373, 112086. [Google Scholar] [CrossRef] [PubMed]
- Own, L.S.; Iqbal, R.; Patel, P.D. Maternal separation alters serotonergic and HPA axis gene expression independent of separation duration in c57bl/6 mice. Brain Res. 2013, 1515, 29–38. [Google Scholar] [CrossRef]
- Vicentic, A.; Francis, D.; Moffett, M.; Lakatos, A.; Rogge, G.; Hubert, G.; Harley, J.; Kuhar, M. Maternal separation alters serotonergic transporter densities and serotonergic 1A receptors in rat brain. Neuroscience 2006, 140, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Hardebo, J.; Edvinsson, L.; Owman, C.; Svendgaard, N.-A. Potentiation and antagonism of serotonin effects on intracranial and extracranial vessels: Possible implications in migraine. Neurology 1978, 28, 64. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M.J.; Hartig, P.R.; Hoffman, B.J. Serotonin 5-HT2C receptor stimulates cyclic GMP formation in choroid plexus. J. Neurochem. 1995, 64, 199–205. [Google Scholar] [CrossRef]
- Hoyer, D.; Clarke, D.E.; Fozard, J.R.; Hartig, P.R.; Martin, G.R.; Mylecharane, E.J.; Saxena, P.R.; Humphrey, P. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (Serotonin). Pharmacol. Rev. 1994, 46, 157–203. [Google Scholar]
- Barnes, N.M.; Ahern, G.P.; Becamel, C.; Bockaert, J.; Camilleri, M.; Chaumont-Dubel, S.; Claeysen, S.; Cunningham, K.A.; Fone, K.C.; Gershon, M. International Union of Basic and Clinical Pharmacology. CX. Classification of receptors for 5-hydroxytryptamine; pharmacology and function. Pharmacol. Rev. 2021, 73, 310–520. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, L.R.; Paleolog, E.M.; Williams, R.O. Kynurenine metabolism in health and disease. Amino Acids 2011, 41, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Moloney, G.M.; O’Leary, O.F.; Salvo-Romero, E.; Desbonnet, L.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. Microbial regulation of hippocampal miRNA expression: Implications for transcription of kynurenine pathway enzymes. Behav. Brain Res. 2017, 334, 50–54. [Google Scholar] [CrossRef]
- Chen, J.-J.; Zeng, B.-H.; Li, W.-W.; Zhou, C.-J.; Fan, S.-H.; Cheng, K.; Zeng, L.; Zheng, P.; Fang, L.; Wei, H. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav. Brain Res. 2017, 322, 34–41. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Yi, L.; Luo, J.-F.; Xie, B.-B.; Liu, J.-X.; Wang, J.-Y.; Liu, L.; Wang, P.-X.; Zhou, H.; Dong, Y. α7 nicotinic acetylcholine receptor is a novel mediator of sinomenine anti-inflammation effect in macrophages stimulated by lipopolysaccharide. Shock 2015, 44, 188–195. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Mikkelsen, J.D. The α7 nicotinic acetylcholine receptor ligands methyllycaconitine, NS6740 and GTS-21 reduce lipopolysaccharide-induced TNF-α release from microglia. J. Neuroimmunol. 2012, 251, 65–72. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-W.; Wang, H.-S.; Zeng, J.; Fang, X.; Chen, H.-Y.; Du, J.; Yang, X.-Y. Sodium butyrate inhibits interferon-gamma induced indoleamine 2, 3-dioxygenase expression via STAT1 in nasopharyngeal carcinoma cells. Life Sci. 2013, 93, 509–515. [Google Scholar] [CrossRef]
- Schwarcz, R. The kynurenine pathway of tryptophan degradation as a drug target. Curr. Opin. Pharmacol. 2004, 4, 12–17. [Google Scholar] [CrossRef]
- Choi, S.-C.; Brown, J.; Gong, M.; Ge, Y.; Zadeh, M.; Li, W.; Croker, B.P.; Michailidis, G.; Garrett, T.J.; Mohamadzadeh, M. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 2020, 12, 551. [Google Scholar] [CrossRef]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Wang, Y.; Liu, Z.-Q.; Zhang, X.; Han, R.; Miao, Y.-Z.; Qin, Z.-H. Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-α. Apoptosis 2017, 22, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R.; Aggerbeck, M.; Aggerbeck, L.; Coumoul, X. The aryl hydrocarbon receptor system. Drug Metab. Pers. Ther. 2012, 27, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Hoban, A.E.; Stilling, R.M.; Moloney, G.M.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F.; Clarke, G. Microbial regulation of microRNA expression in the amygdala and prefrontal cortex. Microbiome 2017, 5, 1–11. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.; Guillemin, G. The plasma [kynurenine]/[tryptophan] ratio and indoleamine 2, 3-dioxygenase: Time for appraisal. Int. J. Tryptophan Res. 2019, 12, 1178646919868978. [Google Scholar] [CrossRef]
- Colliou, N.; Ge, Y.; Sahay, B.; Gong, M.; Zadeh, M.; Owen, J.L.; Neu, J.; Farmerie, W.G.; Alonzo, F.; Liu, K. Commensal Propionibacterium strain UF1 mitigates intestinal inflammation via Th17 cell regulation. J. Clin. Investig. 2017, 127, 3970–3986. [Google Scholar] [CrossRef]
- Valladares, R.; Bojilova, L.; Potts, A.H.; Cameron, E.; Gardner, C.; Lorca, G.; Gonzalez, C.F. Lactobacillus johnsonii inhibits indoleamine 2, 3-dioxygenase and alters tryptophan metabolite levels in BioBreeding rats. FASEB J. 2013, 27, 1711–1720. [Google Scholar] [CrossRef]
- Neufeld, K.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan metabolism by gut microbiome and gut-brain-axis: An in silico analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.M.; Pacheco, A.R.; Henrick, B.M.; Taft, D.; Xu, G.; Huda, M.N.; Mishchuk, D.; Goodson, M.L.; Slupsky, C.; Barile, D. Indole-3-lactic acid associated with Bifidobacterium-dominated microbiota significantly decreases inflammation in intestinal epithelial cells. BMC Microbiol. 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A.; Stockinger, B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009, 30, 447–454. [Google Scholar] [CrossRef]
- Liu, Y.; Tran, D.Q.; Fatheree, N.Y.; Marc Rhoads, J. Lactobacillus reuteri DSM 17938 differentially modulates effector memory T cells and Foxp3+ regulatory T cells in a mouse model of necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G177–G186. [Google Scholar] [CrossRef] [PubMed]
- Rouse, M.; Singh, N.P.; Nagarkatti, P.S.; Nagarkatti, M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br. J. Pharmacol. 2013, 169, 1305–1321. [Google Scholar] [CrossRef]
- Romani, L.; Zelante, T.; Luca, A.D.; Iannitti, R.G.; Moretti, S.; Bartoli, A.; Aversa, F.; Puccetti, P. Microbiota control of a tryptophan—AhR pathway in disease tolerance to fungi. Eur. J. Immunol. 2014, 44, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Behr, C.; Kamp, H.; Fabian, E.; Krennrich, G.; Mellert, W.; Peter, E.; Strauss, V.; Walk, T.; Rietjens, I.; Van Ravenzwaay, B. Gut microbiome-related metabolic changes in plasma of antibiotic-treated rats. Arch. Toxicol. 2017, 91, 3439–3454. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Konopelski, P.; Konop, M.; Gawrys-Kopczynska, M.; Podsadni, P.; Szczepanska, A.; Ufnal, M. Indole-3-propionic acid, a tryptophan-derived bacterial metabolite, reduces weight gain in rats. Nutrients 2019, 11, 591. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, C.; Zhao, D.; Wang, C.; Li, C. Dietary Proteins Regulate Serotonin Biosynthesis and Catabolism by Specific Gut Microbes. J. Agric. Food Chem. 2020, 68, 5880–5890. [Google Scholar] [CrossRef]
- Hata, T.; Asano, Y.; Yoshihara, K.; Kimura-Todani, T.; Miyata, N.; Zhang, X.-T.; Takakura, S.; Aiba, Y.; Koga, Y.; Sudo, N. Regulation of gut luminal serotonin by commensal microbiota in mice. PLoS ONE 2017, 12, e0180745. [Google Scholar] [CrossRef]
- Caldwell, H.D.; Wood, H.; Crane, D.; Bailey, R.; Jones, R.B.; Mabey, D.; Maclean, I.; Mohammed, Z.; Peeling, R.; Roshick, C. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J. Clin. Investig. 2003, 111, 1757–1769. [Google Scholar] [CrossRef]
- Gershon, M.D.; Chalazonitis, A.; Rothman, T.P. From neural crest to bowel: Development of the enteric nervous system. J. Neurobiol. 1993, 24, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Stasi, C.; Sadalla, S.; Milani, S. The relationship between the serotonin metabolism, gut-microbiota and the gut-brain axis. Curr. Drug Metab. 2019, 20, 646–655. [Google Scholar] [CrossRef]
- Takaki, M.; Goto, K.; Kawahara, I. The 5-hydroxytryptamine 4 receptor agonist-induced actions and enteric neurogenesis in the gut. J. Neurogastroenterol. Motil. 2014, 20, 17. [Google Scholar] [CrossRef]
- Masood, J. Neurotransmission Modulates Neurogenesis within the Postnatal Enteric Nervous System. Ph.D. Thesis, Queen’s University, Kingston, ON, Canada, 25 August 2015. [Google Scholar]
- Liu, M.-T.; Kuan, Y.-H.; Wang, J.; Hen, R.; Gershon, M.D. 5-HT4 receptor-mediated neuroprotection and neurogenesis in the enteric nervous system of adult mice. J. Neurosci. 2009, 29, 9683–9699. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chalazonitis, A.; Huang, Y.-Y.; Mann, J.J.; Margolis, K.G.; Yang, Q.M.; Kim, D.O.; Côté, F.; Mallet, J.; Gershon, M.D. Essential roles of enteric neuronal serotonin in gastrointestinal motility and the development/survival of enteric dopaminergic neurons. J. Neurosci. 2011, 31, 8998–9009. [Google Scholar] [CrossRef]
- Waider, J.; Proft, F.; Langlhofer, G.; Asan, E.; Lesch, K.-P.; Gutknecht, L. GABA concentration and GABAergic neuron populations in limbic areas are differentially altered by brain serotonin deficiency in Tph2 knockout mice. Histochem. Cell Biol. 2013, 139, 267–281. [Google Scholar] [CrossRef]
- Ogbonnaya, E.S.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F.; O’Leary, O.F. Adult hippocampal neurogenesis is regulated by the microbiome. Biol. Psychiatry 2015, 78, e7–e9. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Hein, A.M.; O’Banion, M.K.; DiCicco-Bloom, E.; Opanashuk, L.A. Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 2013, 125, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Russi, P.; Lombardi, G.; Beni, M.; Carla, V. Presence of kynurenic acid in the mammalian brain. J. Neurochem. 1988, 51, 177–180. [Google Scholar] [CrossRef]
- Fukuwatari, T. Possibility of amino acid treatment to prevent the psychiatric disorders via modulation of the production of tryptophan metabolite kynurenic acid. Nutrients 2020, 12, 1403. [Google Scholar] [CrossRef]
- Sodhi, M.; Sanders-Bush, E. Serotonin and brain development. Int. Rev. Neurobiol. 2004, 59, 111–174. [Google Scholar] [PubMed]
- Lee, J.; Venna, V.R.; Durgan, D.J.; Shi, H.; Hudobenko, J.; Putluri, N.; Petrosino, J.; McCullough, L.D.; Bryan, R.M. Young versus aged microbiota transplants to germ-free mice: Increased short-chain fatty acids and improved cognitive performance. Gut Microbes 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Misra, S.; Medhi, B. Role of probiotics as memory enhancer. Indian J. Pharmacol. 2013, 45, 311. [Google Scholar]
- Romo-Araiza, A.; Gutiérrez-Salmeán, G.; Galván, E.J.; Hernández-Frausto, M.; Herrera-López, G.; Romo-Parra, H.; García-Contreras, V.; Fernández-Presas, A.M.; Jasso-Chávez, R.; Borlongan, C.V. Probiotics and prebiotics as a therapeutic strategy to improve memory in a model of middle-aged rats. Front. Aging Neurosci. 2018, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yang, R.; Wang, W.; Qi, L.; Huang, T. Associations between gut microbiota and Alzheimer’s disease, major depressive disorder, and schizophrenia. J. Neuroinflamm. 2020, 17, 1–9. [Google Scholar] [CrossRef]
- He, Y.; Li, B.; Sun, D.; Chen, S. Gut microbiota: Implications in Alzheimer’s disease. J. Clin. Med. 2020, 9, 2042. [Google Scholar] [CrossRef] [PubMed]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Wu, H.-Q.; Potter, M.C.; Elmer, G.I.; Pellicciari, R.; Schwarcz, R. Fluctuations in endogenous kynurenic acid control hippocampal glutamate and memory. Neuropsychopharmacology 2011, 36, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Chyan, Y.-J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer β-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef]
- Bendheim, P.E.; Poeggeler, B.; Neria, E.; Ziv, V.; Pappolla, M.A.; Chain, D.G. Development of indole-3-propionic acid (OXIGON™) for alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 213–217. [Google Scholar] [CrossRef]
- Campos-Acuña, J.; Elgueta, D.; Pacheco, R. T-cell-driven inflammation as a mediator of the gut-brain axis involved in Parkinson’s disease. Front. Immunol. 2019, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Lubomski, M.; Tan, A.H.; Lim, S.-Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2019, 267, 1–17. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X. Tryptophan Metabolites Are Associated with Symptoms and Nigral Pathology in Parkinson’s Disease. Mov. Disord. 2020, 35, 2028–2037. [Google Scholar] [CrossRef]
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- González-Sánchez, M.; Jiménez, J.; Narváez, A.; Antequera, D.; Llamas-Velasco, S.; Martín, A.H.-S.; Arjona, J.A.M.; Munain, A.L.D.; Bisa, A.L.; Marco, M. Kynurenic acid levels are increased in the CSF of Alzheimer’s disease patients. Biomolecules 2020, 10, 571. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Pearson, S.; Reynolds, G. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.; Demel, U.; Fuchs, D. Degradation of tryptophan in neurodegenerative disorders. In Tryptophan, Serotonin, and Melatonin; Springer: Berlin/Heidelberg, Germany, 1999; pp. 133–138. [Google Scholar]
- Khaspekov, L.; Lapin, I.; Ryzhov, I.; Viktorov, I. Effects of antagonists of exciting amino acids on the neuro-destructive effect of quinolinic acid in vitro in comparison with their anticonvulsive action in situ. Biull. Eksp. Biol. Med. 1989, 108, 190–193. [Google Scholar] [CrossRef]
- Chen, C.-M. Mitochondrial dysfunction, metabolic deficits, and increased oxidative stress in Huntington’s disease. Chang. Gung Med. J. 2011, 34, 135–152. [Google Scholar] [PubMed]
- Torkildsen, Ø.; Myhr, K.M.; Bø, L. Disease-modifying treatments for multiple sclerosis—A review of approved medications. Eur. J. Neurol. 2016, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; Von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The role of the aryl hydrocarbon receptor (AHR) in immune and inflammatory diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Franco, N.F.; Ng, M.L.; Pai, S.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front. Immunol. 2016, 7, 246. [Google Scholar] [CrossRef]
- Sundaram, G.; Lim, C.K.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression. J. Neuroinflamm. 2020, 17, 1–14. [Google Scholar] [CrossRef]
- Hajsl, M.; Hlavackova, A.; Broulikova, K.; Sramek, M.; Maly, M.; Dyr, J.E.; Suttnar, J. Tryptophan metabolism, inflammation, and oxidative stress in patients with neurovascular disease. Metabolites 2020, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Darlington, L.; Mackay, G.; Forrest, C.; Stoy, N.; George, C.; Stone, T. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef]
- Wigner, P.; Saluk-Bijak, J.; Synowiec, E.; Miller, E.; Sliwinski, T.; Cichon, N.; Bijak, M. Variation of Genes Encoding Tryptophan Catabolites Pathway Enzymes in Stroke. J. Clin. Med. 2019, 8, 2133. [Google Scholar] [CrossRef] [PubMed]
- Winek, K.; Dirnagl, U.; Meisel, A. The gut microbiome as therapeutic target in central nervous system diseases: Implications for stroke. Neurotherapeutics 2016, 13, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Roth, S.; Llovera, G.; Sadler, R.; Garzetti, D.; Stecher, B.; Dichgans, M.; Liesz, A. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci. 2016, 36, 7428–7440. [Google Scholar] [CrossRef]
- Han, D.; Liu, H.; Gao, Y. The role of peripheral monocytes and macrophages in ischemic stroke. Neurol. Sci. 2020, 41, 1–19. [Google Scholar] [CrossRef]
- Yan, K.; Zhang, R.; Sun, C.; Chen, L.; Li, P.; Liu, Y.; Peng, L.; Sun, H.; Qin, K.; Chen, F. Bone marrow-derived mesenchymal stem cells maintain the resting phenotype of microglia and inhibit microglial activation. PLoS ONE 2013, 8, e84116. [Google Scholar]
- Durgan, D.J.; Lee, J.; McCullough, L.D.; Bryan, R.M., Jr. Examining the role of the microbiota-gut-brain axis in stroke. Stroke 2019, 50, 2270–2277. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Hemarajata, P.; Versalovic, J. The human gut microbiome and body metabolism: Implications for obesity and diabetes. Clin. Chem. 2013, 59, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Shikata, F.; Shimada, K.; Sato, H.; Ikedo, T.; Kuwabara, A.; Furukawa, H.; Korai, M.; Kotoda, M.; Yokosuka, K.; Makino, H. Potential influences of gut microbiota on the formation of intracranial aneurysm. Hypertension 2019, 73, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Emilsson, K.; Hardebo, J.E.; Nystedt, S.; Owman, C. Uptake and release of serotonin in rat cerebrovascular nerves after subarachnoid hemorrhage. Stroke 1992, 23, 54–61. [Google Scholar] [CrossRef]
- Lincoln, J. Innervation of cerebral arteries by nerves containing 5-hydroxytryptamine and noradrenaline. Pharmacol. Ther. 1995, 68, 473–501. [Google Scholar] [CrossRef]
- Chima, J.; Mullaguri, N.; Fan, T.; George, P.; Newey, C.R. Cyproheptadine in the treatment of reversible cerebral vasoconstriction syndrome. Acta Neurol. 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mulak, A.; Paradowski, L. Migraine and irritable bowel syndrome. Neurol. Neurochir. Pol. 2005, 39, S55–S60. [Google Scholar] [PubMed]
- Cole, J.A.; Rothman, K.J.; Cabral, H.J.; Zhang, Y.; Farraye, F.A. Migraine, fibromyalgia, and depression among people with IBS: A prevalence study. BMC Gastroenterol. 2006, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schwedt, T.J.; Chiang, C.-C.; Chong, C.D.; Dodick, D.W. Functional MRI of migraine. Lancet Neurol. 2015, 14, 81–91. [Google Scholar] [CrossRef]
- Vikelis, M.; Mitsikostas, D.D. The role of glutamate and its receptors in migraine. CNS Neurol. Disord. Drug Targets 2007, 6, 251–257. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Q.; Wang, A.; Lin, Z. Structural and functional characterization of the gut microbiota in elderly women with migraine. Front. Cell. Infect. Microbiol. 2020, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Arzani, M.; Jahromi, S.R.; Ghorbani, Z.; Vahabizad, F.; Martelletti, P.; Ghaemi, A.; Sacco, S.; Togha, M. Gut-brain Axis and migraine headache: A comprehensive review. J. Headache Pain 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Hinson, H.E.; Rowell, S.; Schreiber, M. Clinical evidence of inflammation driving secondary brain injury: A systematic review. J. Trauma Acute Care Surg. 2015, 78, 184. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef]
- Zhu, C.S.; Grandhi, R.; Patterson, T.T.; Nicholson, S.E. A review of traumatic brain injury and the gut microbiome: Insights into novel mechanisms of secondary brain injury and promising targets for neuroprotection. Brain Sci. 2018, 8, 113. [Google Scholar] [CrossRef]
- Zhang, Z.; Rasmussen, L.; Saraswati, M.; Koehler, R.C.; Robertson, C.; Kannan, S. Traumatic injury leads to inflammation and altered tryptophan metabolism in the juvenile rabbit brain. J. Neurotrauma 2019, 36, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Helmy, A.; Carpenter, K.L.; Menon, D.K.; Pickard, J.D.; Hutchinson, P.J. The cytokine response to human traumatic brain injury: Temporal profiles and evidence for cerebral parenchymal production. J. Cereb. Blood Flow Metab. 2011, 31, 658–670. [Google Scholar] [CrossRef]
- Sinz, E.H.; Kochanek, P.M.; Heyes, M.P.; Wisniewski, S.R.; Bell, M.J.; Clark, R.S.; DeKosky, S.T.; Blight, A.R.; Marion, D.W. Quinolinic acid is increased in CSF and associated with mortality after traumatic brain injury in humans. J. Cereb. Blood Flow Metab. 1998, 18, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Plane, J.M.; Shen, Y.; Pleasure, D.E.; Deng, W. Prospects for minocycline neuroprotection. Arch. Neurol. 2010, 67, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.T.; Shanahan, F.; Dinan, T.T.; Cryan, J.T. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.-F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Borre, Y.; O’Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef]
- Evrensel, A.; Ünsalver, B.Ö.; Ceylan, M.E. Immune-Kynurenine Pathways and the Gut Microbiota-Brain Axis in Anxiety Disorders. Anxiety Disord. 2020, 1191, 155–167. [Google Scholar]
- Dostal, C.R.; Sulzer, M.C.; Kelley, K.W.; Freund, G.G.; McCusker, R.H. Glial and tissue-specific regulation of Kynurenine Pathway dioxygenases by acute stress of mice. Neurobiol. Stress 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.B.; Melas, P.A.; Wegener, G.; Mathé, A.A.; Lavebratt, C. Antidepressant-like effect of sodium butyrate is associated with an increase in TET1 and in 5-hydroxymethylation levels in the Bdnf gene. Int. J. Neuropsychopharmacol. 2015, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Fernandes, I.; Meireles, M.; Faria, A.; Spencer, J.P.; Mateus, N.; Calhau, C. Gut microbiota modulation accounts for the neuroprotective properties of anthocyanins. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Galaktionova, D.I.; Gareeva, A.; Khusnutdinova, E.; Nasedkina, T. The association of polymorphisms in SLC18A1, TPH1 and RELN genes with risk of paranoid schizophrenia. Mol. Biol. 2014, 48, 629–639. [Google Scholar] [CrossRef]
- Brown, G.L.; Ebert, M.H.; Goyer, P.F.; Jimerson, D.C.; Klein, W.J.; Bunney, W.E.; Goodwin, F.K. Aggression, suicide, and serotonin: Relationships of CSF amine metabolites. Am. J. Psychiatry 1982, 139, 741–746. [Google Scholar] [PubMed]
- Erhardt, S.; Blennow, K.; Nordin, C.; Skogh, E.; Lindström, L.H.; Engberg, G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci. Lett. 2001, 313, 96–98. [Google Scholar] [CrossRef]
- Chamberlain, B.; Ervin, F.R.; Pihl, R.O.; Young, S.N. The effect of raising or lowering tryptophan levels on aggression in vervet monkeys. Pharmacol. Biochem. Behav. 1987, 28, 503–510. [Google Scholar] [CrossRef]
- DeNapoli, J.S.; Dodman, N.H.; Shuster, L.; Rand, W.M.; Gross, K.L. Effect of dietary protein content and tryptophan supplementation on dominance aggression, territorial aggression, and hyperactivity in dogs. J. Am. Vet. Med. Assoc. 2000, 217, 504–508. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Canetta, S.; Brown, A. Prenatal infection, maternal immune activation, and risk for schizophrenia. Transl. Neurosci. 2012, 3, 320–327. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Meehan, C.L.; Harms, L.R.; Michie, P.T.; Bigland, M.J.; Smith, D.W.; Jobling, P.; Hodgson, D.M.; Tooney, P.A. Increased complement component 4 (C4) gene expression in the cingulate cortex of rats exposed to late gestation immune activation. Schizophr. Res. 2018, 199, 442–444. [Google Scholar] [CrossRef]
- Kałużna-Czaplińska, J.; Jóźwik-Pruska, J.; Chirumbolo, S.; Bjørklund, G. Tryptophan status in autism spectrum disorder and the influence of supplementation on its level. Metab. Brain Dis. 2017, 32, 1585–1593. [Google Scholar] [CrossRef]
- James, S.J.; Rose, S.; Melnyk, S.; Jernigan, S.; Blossom, S.; Pavliv, O.; Gaylor, D.W. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009, 23, 2374–2383. [Google Scholar] [CrossRef]
- Vuong, H.E.; Hsiao, E.Y. Emerging roles for the gut microbiome in autism spectrum disorder. Biol. Psychiatry 2017, 81, 411–423. [Google Scholar] [CrossRef]
- Iglesias-Vázquez, L.; Van Ginkel Riba, G.; Arija, V.; Canals, J. Composition of gut microbiota in children with autism spectrum disorder: A systematic review and meta-analysis. Nutrients 2020, 12, 792. [Google Scholar] [CrossRef]
- Li, Q.; Han, Y.; Dy, A.B.C.; Hagerman, R.J. The gut microbiota and autism spectrum disorders. Front. Cell. Neurosci. 2017, 11, 120. [Google Scholar] [CrossRef]
- Camilleri, M.; Andrews, C.N.; Bharucha, A.E.; Carlson, P.J.; Ferber, I.; Stephens, D.; Smyrk, T.C.; Urrutia, R.; Aerssens, J.; Thielemans, L. Alterations in expression of p11 and SERT in mucosal biopsy specimens of patients with irritable bowel syndrome. Gastroenterology 2007, 132, 17–25. [Google Scholar] [CrossRef]
- Dunlop, S.P.; Coleman, N.S.; Blackshaw, E.; Perkins, A.C.; Singh, G.; Marsden, C.A.; Spiller, R.C. Abnormalities of 5-hydroxytryptamine metabolism in irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 2005, 3, 349–357. [Google Scholar] [CrossRef]
- Kohen, R.; Jarrett, M.E.; Cain, K.C.; Jun, S.-E.; Navaja, G.P.; Symonds, S.; Heitkemper, M.M. The serotonin transporter polymorphism rs25531 is associated with irritable bowel syndrome. Dig. Dis. Sci. 2009, 54, 2663–2670. [Google Scholar] [CrossRef]
- Clarke, G.; Fitzgerald, P.; Cryan, J.F.; Cassidy, E.M.; Quigley, E.M.; Dinan, T.G. Tryptophan degradation in irritable bowel syndrome: Evidence of indoleamine 2, 3-dioxygenase activation in a male cohort. BMC Gastroenterol. 2009, 9, 1–7. [Google Scholar] [CrossRef]
- Clarke, G.; McKernan, D.P.; Gaszner, G.; Quigley, E.M.; Cryan, J.F.; Dinan, T.G. A distinct profile of tryptophan metabolism along the kynurenine pathway downstream of toll-like receptor activation in irritable bowel syndrome. Front. Pharmacol. 2012, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.M. A role for the gut microbiota in IBS. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.R.; Sherwin, L.B.; Walitt, B.; Melkus, G.D.E.; Henderson, W.A. Neuroimaging the brain-gut axis in patients with irritable bowel syndrome. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 320. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.-J.; Uta, D.; Feng, P.-Y.; Wakita, M.; Shin, M.-C.; Furue, H.; Yoshimura, M. Identification of 5-HT receptor subtypes enhancing inhibitory transmission in the rat spinal dorsal horn in vitro. Mol. Pain 2012, 8, 1744–8069. [Google Scholar] [CrossRef]
- Goldberg, P.A.; Kamm, M.A.; Setti-Carraro, P.; Van der Sijp, J.R.; Roth, C. Modification of visceral sensitivity and pain in irritable bowel syndrome by 5-HT3 antagonism (ondansetron). Digestion 1996, 57, 478–483. [Google Scholar] [CrossRef]
- McKernan, D.; Fitzgerald, P.; Dinan, T.; Cryan, J. The probiotic Bifidobacterium infantis 35624 displays visceral antinociceptive effects in the rat. Neurogastroenterol. Motil. 2010, 22, 1029. [Google Scholar] [CrossRef]
- Ahonen, A.; Kyösola, K.; Penttilä, O. Enterochromaffin cells in macrophages in ulcerative colitis and irritable colon. Ann. Clin. Res. 1976, 8, 1–7. [Google Scholar]
- Buchan, A.; Grant, S.; Brown, J.; Freeman, H. A quantitative study of enteric endocrine cells in celiac sprue. J. Pediatr. Gastroenterol. Nutr. 1984, 3, 665–671. [Google Scholar] [CrossRef]
- Foley, K.F.; Pantano, C.; Ciolino, A.; Mawe, G.M. IFN-γ and TNF-α decrease serotonin transporter function and expression in Caco2 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G779–G784. [Google Scholar] [CrossRef]
- Cawthon, C.R.; Claire, B. Gut bacteria interaction with vagal afferents. Brain Res. 2018, 1693, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Ghia, J.E.; Li, N.; Wang, H.; Collins, M.; Deng, Y.; El–Sharkawy, R.T.; Côté, F.; Mallet, J.; Khan, W.I. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology 2009, 137, 1649–1660. [Google Scholar] [CrossRef]
- Roth, W.H.; Cai, A.; Zhang, C.; Chen, M.L.; Merkler, A.E.; Kamel, H. Gastrointestinal Disorders and Risk of First-Ever Ischemic Stroke. Stroke 2020, 51, 3577–3583. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. https://doi.org/10.3390/ijms22062973
Roth W, Zadeh K, Vekariya R, Ge Y, Mohamadzadeh M. Tryptophan Metabolism and Gut-Brain Homeostasis. International Journal of Molecular Sciences. 2021; 22(6):2973. https://doi.org/10.3390/ijms22062973
Chicago/Turabian StyleRoth, William, Kimia Zadeh, Rushi Vekariya, Yong Ge, and Mansour Mohamadzadeh. 2021. "Tryptophan Metabolism and Gut-Brain Homeostasis" International Journal of Molecular Sciences 22, no. 6: 2973. https://doi.org/10.3390/ijms22062973
APA StyleRoth, W., Zadeh, K., Vekariya, R., Ge, Y., & Mohamadzadeh, M. (2021). Tryptophan Metabolism and Gut-Brain Homeostasis. International Journal of Molecular Sciences, 22(6), 2973. https://doi.org/10.3390/ijms22062973