
Targeting Intercellular Communication in the Bone Microenvironment to Prevent Disseminated Tumor Cell Escape from Dormancy and Bone Metastatic Tumor Growth


Abstract
1. Bone Metastasis
2. Current Therapy for Bone Metastases
3. Inter-Cellular Regulation of Bone Metastasis Growth
3.1. Role of Bone Remodeling in Bone Metastatic Tumor Growth
3.2. Angiogenesis in Bone Metastasis
3.3. Immune Cell Function in Bone Metastasis
3.3.1. T-Cells
3.3.2. Macrophages
3.3.3. MDSC
4. Rethinking Therapy for Bone Metastases
4.1. NR2F1 as an Inducer of Dormancy
4.2. The Gas6-Axl-TGF-β Axis
4.3. CXCL12/CXCR4 Signaling
4.4. Integrin-FAK Signaling
4.5. The IL-6-STAT3-JAK Signaling Axis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kopp, H.-G.; Avecilla, S.T.; Hooper, A.T.; Rafii, S. The Bone Marrow Vascular Niche: Home of HSC Differentiation and Mobilization. Physiology (Bethesda) 2005, 20, 349–356. [Google Scholar] [CrossRef]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell. Mol. Immunol. 2011, 9, 11–19. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shen, Y. Study on the distribution features of bone metastases in prostate cancer. Nucl. Med. Commun. 2012, 33, 379–383. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, Z. Clinical Characteristics and Prognostic Factors in Bone Metastases from Lung Cancer. Med Sci. Monit. 2017, 23, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.; Barlogie, B.; Haessler, J.; Tricot, G.; Anaissie, E.; Shaughnessy, J.D., Jr.; Epstein, J.; Van Hemert, R.; Erdem, E.; Hoering, A.; et al. Magnetic Resonance Imaging in Multiple Myeloma: Diagnostic and Clinical Implications. J. Clin. Oncol. 2007, 25, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Ohnuma, N.; Iwai, J.; Yoshida, H.; Takahashi, H.; Maie, M.; Etoh, T.; Kawamura, K. Bone Marrow Metastasis of Neuroblastoma Analyzed by MRI and Its Influence on Prognosis. Med Pediatr. Oncol. 1995, 24, 292–299. [Google Scholar] [CrossRef]
- Chen, S.-C.; Kuo, P.-L. Bone Metastasis from Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 987. [Google Scholar] [CrossRef] [PubMed]
- Iñiguez-Ariza, N.M.; Bible, K.C.; Clarke, B.L. Bone metastases in thyroid cancer. J. Bone Oncol. 2020, 21, 100282. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Allocca, G.; Hughes, R.; Wang, N.; Brown, H.K.; Ottewell, P.D.; Brown, N.J.; Holen, I. The bone metastasis niche in breast cancer: Potential overlap with the haematopoietic stem cell niche in vivo. J. Bone Oncol. 2019, 17, 100244. [Google Scholar] [CrossRef]
- Tsuya, A.; Kurata, T.; Tamura, K.; Fukuoka, M. Skeletal metastases in non-small cell lung cancer: A retrospective study. Lung Cancer 2007, 57, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Calhoun, F.W. The distribution of skeletal metastases in breast and pulmonary cancer: Concise communication. J. Nucl. Med. 1981, 22, 594–597. [Google Scholar] [PubMed]
- Wang, C.-Y.; Wu, G.-Y.; Shen, M.-J.; Cui, K.-W.; Shen, Y. Comparison of distribution characteristics of metastatic bone lesions between breast and prostate carcinomas. Oncol. Lett. 2012, 5, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Xue, X.; Hu, C.; Xu, H.; Kou, D.; Li, R.; Li, M. Comparison of Clinicopathological Features and Prognosis in Triple-Negative and Non-Triple Negative Breast Cancer. J. Cancer 2016, 7, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Ragel, B.T.; Reddington, J.A.; Mendez, G.A.; Ching, A.; Kubicky, C.D.; Klimo, P. Imaging characteristic analysis of metastatic spine lesions from breast, prostate, lung, and renal cell carcinomas for surgical planning: Osteolytic versus osteoblastic. Surg. Neurol. Int. 2016, 7, 361–S365. [Google Scholar] [CrossRef] [PubMed]
- Keller, E.T.; Brown, J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J. Cell. Biochem. 2003, 91, 718–729. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Hall, C.L.; Bafico, A.; Dai, J.; Aaronson, S.A.; Keller, E.T. Prostate Cancer Cells Promote Osteoblastic Bone Metastases through Wnts. Cancer Res. 2005, 65, 7554–7560. [Google Scholar] [CrossRef]
- Roudier, M.P.; Morrissey, C.; True, L.D.; Higano, C.S.; Vessella, R.L.; Ott, S.M. Histopathological Assessment of Prostate Cancer Bone Osteoblastic Metastases. J. Urol. 2008, 180, 1154–1160. [Google Scholar] [CrossRef]
- Park, B.K.; Zhang, H.; Zeng, Q.; Dai, J.; Keller, E.T.; Giordano, T.J.; Gu, K.; Shah, V.R.; Pei, L.; Zarbo, R.J.; et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 2007, 13, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef]
- Aielli, F.; Ponzetti, M.; Rucci, N. Bone Metastasis Pain, from the Bench to the Bedside. Int. J. Mol. Sci. 2019, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Kosteva, J.; Langer, C. The changing landscape of the medical management of skeletal metastases in nonsmall cell lung cancer. Curr. Opin. Oncol. 2008, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Chow, E.; Zhang, L.; Velikova, G.; Bezjak, A.; Wu, J.; Barton, M.; Sezer, O.; Eek, R.; Shafiq, J.; et al. Patients’ and health care professionals’ evaluation of health-related quality of life issues in bone metastases. Eur. J. Cancer 2009, 45, 2510–2518. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical Features of Metastatic Bone Disease and Risk of Skeletal Morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef]
- Percival, R.C.; Yates, A.J.; Gray, R.E.; Galloway, J.; Rogers, K.; Neal, F.E.; Kanis, J.A. Mechanism of malignant hypercalcaemia in carcinoma of the breast. BMJ 1985, 291, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Diel, I.J.; Kaufmann, M.; Goerner, R.; Costa, S.D.; Kaul, S.; Bastert, G. Detection of tumor cells in bone marrow of patients with primary breast cancer: A prognostic factor for distant metastasis. J. Clin. Oncol. 1992, 10, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Wiedswang, G.; Borgen, E.; Kåresen, R.; Kvalheim, G.; Nesland, J.; Qvist, H.; Schlichting, E.; Sauer, T.; Janbu, J.; Harbitz, T.; et al. Detection of Isolated Tumor Cells in Bone Marrow Is an Independent Prognostic Factor in Breast Cancer. J. Clin. Oncol. 2003, 21, 3469–3478. [Google Scholar] [CrossRef]
- Bidard, F.-C.; Vincent-Salomon, A.; Gomme, S.; Nos, C.; De Rycke, Y.; Thiery, J.P.; Sigal-Zafrani, B.; Mignot, L.; Sastre-Garau, X.; Pierga, J.-Y. Disseminated Tumor Cells of Breast Cancer Patients: A Strong Prognostic Factor for Distant and Local Relapse. Clin. Cancer Res. 2008, 14, 3306–3311. [Google Scholar] [CrossRef]
- Braun, S.; Pantel, K. Micrometastatic bone marrow involvement: Detection and prognostic significance. Med. Oncol. 1999, 16, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Enzmann, T.; Köllermann, J.; Caprano, J.; Riethmüller, G.; Köllermann, M.W. Immunocytochemical monitoring of micrometastatic disease: Reduction of prostate cancer cells in bone marrow by androgen deprivation. Int. J. Cancer 1997, 71, 521–525. [Google Scholar] [CrossRef]
- Pantel, K.; Angstwurm, M.; Riethmüller, G.; Passlick, B.; Izbicki, J.; Thetter, O.; Häussinger, K. Frequency and prognostic significance of isolated tumour cells in bone marrow of patients with non-small-cell lung cancer without overt metastases. Lancet 1996, 347, 649–653. [Google Scholar] [CrossRef]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N. Engl. J. Med. 2005, 353, 793–802. [Google Scholar] [CrossRef]
- Janni, W.; Vogl, F.D.; Wiedswang, G.; Synnestvedt, M.; Fehm, T.; Jückstock, J.; Borgen, E.; Rack, B.; Braun, S.; Sommer, H.; et al. Persistence of Disseminated Tumor Cells in the Bone Marrow of Breast Cancer Patients Predicts Increased Risk for Relapse—A European Pooled Analysis. Clin. Cancer Res. 2011, 17, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.M.; Lange, P.H.; Porter, M.P.; Lin, D.W.; Ellis, W.J.; Gallaher, I.S.; Vessella, R.L. Disseminated Tumor Cells in Prostate Cancer Patients after Radical Prostatectomy and without Evidence of Disease Predicts Biochemical Recurrence. Clin. Cancer Res. 2009, 15, 677–683. [Google Scholar] [CrossRef]
- Ruffato, A.; D’Ovidio, F.; Pilotti, V.; Tazzari, P.; Mattioli, S.; Pileri, S.; Daddi, N. Do bone marrow isolated tumor cells influence long-term survival of non-small cell lung cancer?? Eur. J. Cardio-Thorac. Surg. 2009, 35, 463–468. [Google Scholar] [CrossRef]
- Rud, A.K.; Borgen, E.; Mælandsmo, G.M.; Flatmark, K.; Le, H.; Josefsen, D.; Solvoll, I.; Schirmer, C.B.; Helland, Å.; Jørgensen, L.; et al. Clinical significance of disseminated tumour cells in non-small cell lung cancer. Br. J. Cancer 2013, 109, 1264–1270. [Google Scholar] [CrossRef]
- Mansi, J.; Morden, J.; Bliss, J.M.; Neville, M.; Coombes, R.C. Bone marrow micrometastases in early breast cancer–30-year outcome. Br. J. Cancer 2016, 114, 243–247. [Google Scholar] [CrossRef][Green Version]
- Tjensvoll, K.; Nordgård, O.; Skjæveland, M.; Oltedal, S.; Janssen, E.A.M.; Gilje, B. Detection of disseminated tumor cells in bone marrow predict late recurrences in operable breast cancer patients. BMC Cancer 2019, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Wallwiener, M.; Fehm, T.N.; Hahn, M.; Walter, C.B.; Gruber, I.; Brucker, S.Y.; Taran, F.-A. Disseminated tumor cells from the bone marrow of patients with nonmetastatic primary breast cancer are predictive of locoregional relapse. Ann. Oncol. 2015, 26, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Köllermann, J.; Weikert, S.; Schostak, M.; Kempkensteffen, C.; Kleinschmidt, K.; Rau, T.; Pantel, K. Prognostic Significance of Disseminated Tumor Cells in the Bone Marrow of Prostate Cancer Patients Treated with Neoadjuvant Hormone Treatment. J. Clin. Oncol. 2008, 26, 4928–4933. [Google Scholar] [CrossRef] [PubMed]
- Willis, L.; Alarcón, T.; Elia, G.; Jones, J.L.; Wright, N.A.; Tomlinson, I.P.; Graham, T.A.; Page, K.M. Breast Cancer Dormancy Can Be Maintained by Small Numbers of Micrometastases. Cancer Res. 2010, 70, 4310–4317. [Google Scholar] [CrossRef]
- Zincke, H.; Oesterling, J.E.; Blute, M.L.; Bergstralh, E.J.; Myers, R.P.; Barrett, D.M. Long-Term (15 Years) Results After Radical Prostatectomy for Clinically Localized (Stage T2c Or Lower) Prostate Cancer. J. Urol. 1994, 152, 1850–1857. [Google Scholar] [CrossRef]
- Pound, C.R.; Partin, A.W.; Eisenberger, M.A.; Chan, D.W.; Pearson, J.D.; Walsh, P.C. Natural History of Progression After PSA Elevation Following Radical Prostatectomy. JAMA 1999, 281, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, K.J.; Macdonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep Nature of Metastatic Inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Patel, L.R.; Camacho, D.F.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Mechanisms of cancer cell metastasis to the bone: A multistep process. Future Oncol. 2011, 7, 1285–1297. [Google Scholar] [CrossRef]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap Junction–Mediated Import of MicroRNA from Bone Marrow Stromal Cells Can Elicit Cell Cycle Quiescence in Breast Cancer Cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef]
- Van Der Toom, E.E.; Verdone, J.E.; Pienta, K.J. Disseminated tumor cells and dormancy in prostate cancer metastasis. Curr. Opin. Biotechnol. 2016, 40, 9–15. [Google Scholar] [CrossRef]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv. Cancer Res. 2016, 132, 45–71. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Li, S.; Kennedy, M.; Payne, S.; Kennedy, K.; Seewaldt, V.L.; Pizzo, S.V.; Bachelder, R.E. Model of Tumor Dormancy/Recurrence after Short-Term Chemotherapy. PLoS ONE 2014, 9, e98021. [Google Scholar] [CrossRef]
- Moore, N.; Houghton, J.; Lyle, S. Slow-Cycling Therapy-Resistant Cancer Cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Riethdorf, S.; Pantel, M.M.S.R.K. Circulating and disseminated tumour cells—Mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 2017, 14, 155–167. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Ossowski, L.; Rosenbaum, S.K.; Atadja, P.; Gao, L.; Kwon, P.; Trogani, N.; Walker, H.; Hsu, M.; Yeleswarapu, L.; et al. Green Fluorescent Protein Tagging of Extracellular Signal-Regulated Kinase and p38 Pathways Reveals Novel Dynamics of Pathway Activation during Primary and Metastatic Growth. Cancer Res. 2004, 64, 7336–7345. [Google Scholar] [CrossRef] [PubMed]
- Yu-Lee, L.-Y.; Yu, G.; Lee, Y.-C.; Lin, S.-C.; Pan, J.; Pan, T.; Yu, K.-J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII–p38MAPK–pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Kramer, R.H. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene 2009, 29, 1214–1226. [Google Scholar] [CrossRef]
- Endo, H.; Okuyama, H.; Ohue, M.; Inoue, M. Dormancy of Cancer Cells with Suppression of AKT Activity Contributes to Survival in Chronic Hypoxia. PLoS ONE 2014, 9, e98858. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Xu, Z.-L.; Zhao, T.-J.; Ye, L.-H.; Zhang, X.-D. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008, 269, 67–77. [Google Scholar] [CrossRef]
- Attar-Schneider, O.; Zismanov, V.; Drucker, L.; Gottfried, M. Secretome of human bone marrow mesenchymal stem cells: An emerging player in lung cancer progression and mechanisms of translation initiation. Tumor Biol. 2015, 37, 4755–4765. [Google Scholar] [CrossRef] [PubMed]
- Páez, D.; LaBonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.-J. Cancer Dormancy: A Model of Early Dissemination and Late Cancer Recurrence. Clin. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef]
- Coleman, R. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- So, A.; Chin, J.; Fleshner, N.; Saad, F. Management of skeletal-related events in patients with advanced prostate cancer and bone metastases: Incorporating new agents into clinical practice. Can. Urol. Assoc. J. 2012, 6, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, K.S.; Fournier, P.G.; Guise, T.A.; Chirgwin, J.M. Agents Targeting Prostate Cancer Bone Metastasis. Anti-Cancer Agents Med. Chem. 2009, 9, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Fauzee, N.J. Taxanes: Promising anti-cancer drugs. Asian Pac. J. Cancer Prev. APJCP 2011, 12, 837–851. [Google Scholar]
- Brown, H.; Ottewell, P.; Evans, C.; Coleman, R.; Holen, I. A single administration of combination therapy inhibits breast tumour progression in bone and modifies both osteoblasts and osteoclasts. J. Bone Oncol. 2012, 1, 47–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ottewell, P.D.; Woodward, J.K.; Lefley, D.V.; Evans, C.A.; Coleman, R.E.; Holen, I. Anticancer mechanisms of doxorubicin and zoledronic acid in breast cancer tumor growth in bone. Mol. Cancer Ther. 2009, 8, 2821–2832. [Google Scholar] [CrossRef]
- Levasseur, N.; Clemons, M.; Hutton, B.; Shorr, R.; Jacobs, C. Bone-targeted therapy use in patients with bone metastases from lung cancer: A systematic review of randomized controlled trials. Cancer Treat. Rev. 2016, 50, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Sawamoto, K.; Alméciga-Díaz, C.J.; MacKenzie, W.G.; Mason, R.W.; Orii, T.; Tomatsu, S. Development of Bone Targeting Drugs. Int. J. Mol. Sci. 2017, 18, 1345. [Google Scholar] [CrossRef]
- Low, S.A.; Galliford, C.V.; Yang, J.; Low, P.S.; Kopeček, J. Biodistribution of Fracture-Targeted GSK3β Inhibitor-Loaded Micelles for Improved Fracture Healing. Biomacromolecules 2015, 16, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Niikura, N.; Liu, J.; Hayashi, N.; Palla, S.L.; Tokuda, Y.; Hortobagyi, G.N.; Ueno, N.T.; Theriault, R.L. Treatment Outcome and Prognostic Factors for Patients with Bone-Only Metastases of Breast Cancer: A Single-Institution Retrospective Analysis. Oncology 2011, 16, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, J.; Chen, M.; Chen, D.; Ye, S.; Li, X.; Chen, X.; Ren, G.; Yan, S. Sites of synchronous distant metastases and prognosis in prostate cancer patients with bone metastases at initial diagnosis: A population-based study of 16,643 patients. Clin. Transl. Med. 2019, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Kentenich, C.; Janni, W.; Hepp, F.; De Waal, J.; Willgeroth, F.; Sommer, H.; Pantel, K. Lack of Effect of Adjuvant Chemotherapy on the Elimination of Single Dormant Tumor Cells in Bone Marrow of High-Risk Breast Cancer Patients. J. Clin. Oncol. 2000, 18, 80. [Google Scholar] [CrossRef]
- Drageset, V.; Nesland, J.M.; Erikstein, B.; Skovlund, E.; Sommer, H.; Anker, G.; Wist, E.; Lundgren, S.; Bergh, J.; Kvalheim, G. Monitoring of disseminated tumor cells in bone marrow in high-risk breast cancer patients treated with high-dose chemotherapy. Int. J. Cancer 2006, 118, 2877–2881. [Google Scholar] [CrossRef]
- Naumov, G.N.; Townson, J.L.; Macdonald, I.C.; Wilson, S.M.; Bramwell, V.H.; Groom, A.C.; Chambers, A.F. Ineffectiveness of Doxorubicin Treatment on Solitary Dormant Mammary Carcinoma Cells or Late-developing Metastases. Breast Cancer Res. Treat. 2003, 82, 199–206. [Google Scholar] [CrossRef]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 Regulates Prostate Cancer Dormancy through mTOR Inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional Coupling of p38-Induced Up-regulation of BiP and Activation of RNA-Dependent Protein Kinase–Like Endoplasmic Reticulum Kinase to Drug Resistance of Dormant Carcinoma Cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Hain, B.A.; Xu, H.; Wilcox, J.R.; Mutua, D.; Waning, D.L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Van Leeuwen, B.L.; Hartel, R.M.; Jansen, H.W.B.; Kamps, W.A.; Hoekstra, H.J. The effect of chemotherapy on the morphology of the growth plate and metaphysis of the growing skeleton. Eur. J. Surg. Oncol. (EJSO) 2003, 29, 49–58. [Google Scholar] [CrossRef]
- Fan, C.; Georgiou, K.R.; McKinnon, R.A.; Keefe, D.M.K.; Howe, P.R.C.; Xian, C.J. Combination chemotherapy with cyclophosphamide, epirubicin and 5-fluorouracil causes trabecular bone loss, bone marrow cell depletion and marrow adiposity in female rats. J. Bone Miner. Metab. 2015, 34, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hadji, P.; Ziller, M.; Maskow, C.; Albert, U.; Kalder, M. The influence of chemotherapy on bone mineral density, quantitative ultrasonometry and bone turnover in pre-menopausal women with breast cancer. Eur. J. Cancer 2009, 45, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Chakrabarti, A.; Freeman, M.; Biswas, S. Doxorubicin-Mediated Bone Loss in Breast Cancer Bone Metastases Is Driven by an Interplay between Oxidative Stress and Induction of TGFβ. PLoS ONE 2013, 8, e78043. [Google Scholar] [CrossRef]
- Weekes, C.D.; Kuszynski, C.A.; Sharp, J.G. VLA-4 Mediated Adhesion to Bone Marrow Stromal Cells Confers Chemoresistance to Adherent Lymphoma Cells. Leuk. Lymphoma 2001, 40, 631–645. [Google Scholar] [CrossRef]
- Kar, S.; Katti, D.R.; Katti, K.S. Bone interface modulates drug resistance in breast cancer bone metastasis. Colloids Surf. B Biointerfaces 2020, 195, 111224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Lin, S.-C.; Yu, G.; Cheng, C.-J.; Liu, B.; Liu, H.-C.; Hawke, D.H.; Parikh, N.U.; Varkaris, A.; Corn, P.G.; et al. Identification of Bone-Derived Factors Conferring De Novo Therapeutic Resistance in Metastatic Prostate Cancer. Cancer Res. 2015, 75, 4949–4959. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options. J. Bone Oncol. 2019, 15, 100205. [Google Scholar] [CrossRef]
- Green, J.R. Bisphosphonates: Preclinical Review. Oncology 2004, 9, 3–13. [Google Scholar] [CrossRef]
- Hughes, D.E.; Wright, K.R.; Uy, H.L.; Sasaki, A.; Yoneda, T.; Roodman, D.G.; Mundy, G.R.; Boyce, B.F. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J. Bone Miner. Res. 2009, 10, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.J.; Crockett, J.C.; Coxon, F.P.; Mönkkönen, J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone 2011, 49, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Figgitt, D.P. Zoledronic Acid. Drugs 2004, 64, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Polascik, T.J. Bisphosphonates in oncology: Evidence for the prevention of skeletal events in patients with bone metastases. Drug Des. Dev. Ther. 2008, 3, 27–40. [Google Scholar] [CrossRef][Green Version]
- Park, J.H.; Lee, N.K.; Lee, A.S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.-J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab Compared With Zoledronic Acid for the Treatment of Bone Metastases in Patients With Advanced Breast Cancer: A Randomized, Double-Blind Study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F.; Liang, G.; Zeng, X.; Yu, W.; Jiang, Z.; Ma, J.; Zhao, M.; Xiong, M.; Gui, K.; et al. Denosumab versus zoledronic acid for preventing symptomatic skeletal events in Asian postmenopausal women with oestrogen-receptor-positive advanced breast cancer: An outcome analyses with a mean follow-up of 3 years. BMC Musculoskelet. Disord. 2018, 19, 424. [Google Scholar] [CrossRef]
- Imai, H.; Saijo, K.; Yamada, H.; Ohuchi, K.; Okada, Y.; Komine, K.; Takahashi, M.; Takahashi, S.; Takahashi, M.; Shimodaira, H.; et al. Efficacy and safety of denosumab versus zoledronic acid in delaying skeletal-related events in patients with gastrointestinal cancer, pancreas-biliary system cancer, and other rare cancers. J. Bone Oncol. 2017, 6, 37–40. [Google Scholar] [CrossRef][Green Version]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damião, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Lutz, S.; Balboni, T.; Jones, J.; Lo, S.; Petit, J.; Rich, S.E.; Wong, R.; Hahn, C. Palliative radiation therapy for bone metastases: Update of an ASTRO Evidence-Based Guideline. Pr. Radiat. Oncol. 2017, 7, 4–12. [Google Scholar] [CrossRef]
- De Felice, F.; Piccioli, A.; Musio, D.; Tombolini, V. The role of radiation therapy in bone metastases management. Oncotarget 2017, 8, 25691–25699. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Nilsson, D.S.; Heinrich, S.D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Strang, P.; Aksnes, A.; Franzèn, L.; Olivier, P.; Pecking, A.; Staffurth, J.; Vasanthan, S.; Andersson, C.; Bruland, Ø.S. A randomized, dose–response, multicenter phase II study of radium-223 chloride for the palliation of painful bone metastases in patients with castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; Coleman, R.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: Results from a phase 3, double-blind, randomised trial. Lancet Oncol. 2014, 15, 738–746. [Google Scholar] [CrossRef]
- Zenda, S.; Nakagami, Y.; Toshima, M.; Arahira, S.; Kawashima, M.; Matsumoto, Y.; Kinoshita, H.; Satake, M.; Akimoto, T. Strontium-89 (Sr-89) chloride in the treatment of various cancer patients with multiple bone metastases. Int. J. Clin. Oncol. 2013, 19, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Soeharno, H.; Povegliano, L.; Choong, P.F. Multimodal Treatment of Bone Metastasis—A Surgical Perspective. Front. Endocrinol. 2018, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M. Bone remodeling, normal and abnormal: A biological basis for the understanding of cancer-related bone disease and its treatment. Can. J. Oncol. 1995, 5, 1–10. [Google Scholar]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast–osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef]
- Shahi, M.; Peymani, A.; Sahmani, M. Regulation of Bone Metabolism. Rep. Biochem. Mol. Biol. 2017, 5, 73–82. [Google Scholar]
- Kim, J.-M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.-H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Brown, J.E.; Cook, R.J.; Major, P.; Lipton, A.; Saad, F.; Smith, M.R.; Lee, K.-A.; Zheng, M.; Hei, Y.-J.; Coleman, R.E. Bone Turnover Markers as Predictors of Skeletal Complications in Prostate Cancer, Lung Cancer, and Other Solid Tumors. J. Natl. Cancer Inst. 2005, 97, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Major, P.; Lipton, A.; Brown, J.E.; Lee, K.-A.; Smith, M.; Saad, F.; Zheng, M.; Hei, Y.J.; Seaman, J.; et al. Predictive Value of Bone Resorption and Formation Markers in Cancer Patients with Bone Metastases Receiving the Bisphosphonate Zoledronic Acid. J. Clin. Oncol. 2005, 23, 4925–4935. [Google Scholar] [CrossRef] [PubMed]
- Kremer, R.; Li, J.; Camirand, A.; Karaplis, A.C. Parathyroid Hormone Related Protein (PTHrP) in Tumor Progression. Adv. Exp. Med. Biol. 2011, 720, 145–160. [Google Scholar] [CrossRef] [PubMed]
- McCauley, L.K.; Martin, T.J. Twenty-five years of PTHrP progress: From cancer hormone to multifunctional cytokine. J. Bone Miner. Res. 2012, 27, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sun, Y.; Ho, P.W.M.; Chan, A.S.M.; Johnson, J.A.; Pavlos, N.J.; Sims, N.A.; Martin, T.J. Parathyroid Hormone-Related Protein Negatively Regulates Tumor Cell Dormancy Genes in a PTHR1/Cyclic AMP-Independent Manner. Front. Endocrinol. 2018, 9, 241. [Google Scholar] [CrossRef]
- Amizuka, N.; Karaplis, A.C.; Henderson, J.E.; Warshawsky, H.; Lipman, M.L.; Matsuki, Y.; Ejiri, S.; Tanaka, M.; Izumi, N.; Ozawa, H.; et al. Haploinsufficiency of Parathyroid Hormone-Related Peptide (PTHrP) Results in Abnormal Postnatal Bone Development. Dev. Biol. 1996, 175, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.K.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP Receptor in Early Development and Indian Hedgehog--Regulated Bone Growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; He, B.; Jiang, Y.; Kobayashi, T.; Sorocéanu, M.A.; Zhao, J.; Tatsuya, K.; Tong, X.; Amizuka, N.; Gupta, A.; et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J. Clin. Investig. 2005, 115, 2402–2411. [Google Scholar] [CrossRef]
- Ren, G.; Esposito, M.; Kang, Y. Bone metastasis and the metastatic niche. J. Mol. Med. 2015, 93, 1203–1212. [Google Scholar] [CrossRef]
- Pang, X.; Gong, K.; Zhang, X.; Wu, S.; Cui, Y.; Qian, B.-Z. Osteopontin as a multifaceted driver of bone metastasis and drug resistance. Pharmacol. Res. 2019, 144, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Isowa, S.; Shimo, T.; Ibaragi, S.; Kurio, N.; Okui, T.; Matsubara, K.; Hassan, N.M.M.; Kishimoto, K.; Sasaki, A. PTHrP Regulates Angiogenesis and Bone Resorption via VEGF Expression. Anticancer Res. 2010, 30, 2755–2767. [Google Scholar] [PubMed]
- Zhang, Z.; Wang, H.; Ikeda, S.; Fahey, F.; Bielenberg, D.; Smits, P.; Hauschka, P.V. Notch3 in Human Breast Cancer Cell Lines Regulates Osteoblast-Cancer Cell Interactions and Osteolytic Bone Metastasis. Am. J. Pathol. 2010, 177, 1459–1469. [Google Scholar] [CrossRef]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-Derived Jagged1 Promotes Osteolytic Bone Metastasis of Breast Cancer by Engaging Notch Signaling in Bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef]
- Zheng, H.; Bae, Y.; Kasimir-Bauer, S.; Tang, R.; Chen, J.; Ren, G.; Yuan, M.; Esposito, M.; Li, W.; Wei, Y.; et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell 2017, 32, 731–747. [Google Scholar] [CrossRef]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clément-Lacroix, P.; Clézardin, P. Tumor αvβ3 Integrin Is a Therapeutic Target for Breast Cancer Bone Metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Hedgehog Signaling Induced by Breast Cancer Cells Promotes Osteoclastogenesis and Osteolysis*. J. Biol. Chem. 2011, 286, 9612–9622. [Google Scholar] [CrossRef]
- McKee, M.D.; Nanci, A. Osteopontin: An Interfacial Extracellular Matrix Protein in Mineralized Tissues. Connect. Tissue Res. 1996, 35, 197–205. [Google Scholar] [CrossRef]
- Tilli, T.M.; Mello, K.D.; Ferreira, L.B.; Matos, A.R.; Accioly, M.T.S.; Faria, P.A.; Bellahcène, A.; Castronovo, V.; Gimba, E.R. Both osteopontin-c and osteopontin-b splicing isoforms exert pro-tumorigenic roles in prostate cancer cells. Prostate 2012, 72, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Berendsen, A.D.; Jia, S.; Lotinun, S.; Baron, R.; Ferrara, N.; Olsen, B.R. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J. Clin. Investig. 2012, 122, 3101–3113. [Google Scholar] [CrossRef]
- Dai, J.; Kitagawa, Y.; Zhang, J.; Yao, Z.; Mizokami, A.; Cheng, S.; Nör, J.; McCauley, L.K.; Taichman, R.S.; Keller, E.T. Vascular Endothelial Growth Factor Contributes to the Prostate Cancer-Induced Osteoblast Differentiation Mediated by Bone Morphogenetic Protein. Cancer Res. 2004, 64, 994–999. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Dai, J.; Zhang, J.; Keller, J.M.; Nor, J.; Yao, Z.; Keller, E.T. Vascular Endothelial Growth Factor Contributes to Prostate Cancer–Mediated Osteoblastic Activity. Cancer Res. 2005, 65, 10921–10929. [Google Scholar] [CrossRef]
- Dewerchin, M.; Carmeliet, P. PlGF: A Multitasking Cytokine with Disease-Restricted Activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Maes, C.; Coenegrachts, L.; Stockmans, I.; Daci, E.; Luttun, A.; Petryk, A.; Gopalakrishnan, R.; Moermans, K.; Smets, N.; Verfaillie, C.M.; et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J. Clin. Investig. 2006, 116, 1230–1242. [Google Scholar] [CrossRef]
- Zhou, X.; Barsky, L.W.; Adams, G.B. Placental Growth Factor Expression Is Required for Bone Marrow Endothelial Cell Support of Primitive Murine Hematopoietic Cells. PLoS ONE 2013, 8, e67861. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coenegrachts, L.; Maes, C.; Torrekens, S.; Van Looveren, R.; Mazzone, M.; Guise, T.A.; Bouillon, R.; Stassen, J.-M.; Carmeliet, P.; Carmeliet, G. Anti–Placental Growth Factor Reduces Bone Metastasis by Blocking Tumor Cell Engraftment and Osteoclast Differentiation. Cancer Res. 2010, 70, 6537–6547. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; De Mol, M.; et al. Anti-PlGF Inhibits Growth of VEGF(R)-Inhibitor-Resistant Tumors without Affecting Healthy Vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef]
- McDonnell, C.; Hill, A.; McNamara, D.; Walsh, T.; Bouchier-Hayes, D. REVIEWS—Tumour micrometastases: The influence of angiogenesis. Eur. J. Surg. Oncol. (EJSO) 2000, 26, 105–115. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef]
- Sherwood, L.M.; Parris, E.E.; Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Fox, S.B.; Gatter, K.C.; Leek, R.D.; Harris, A.L.; Bliss, J.; Mansi, J.L.; Gusterson, B. Association of tumor angiogenesis with bone marrow micrometastases in breast cancer patients. J. Natl. Cancer Inst. 1997, 89, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Aalinkeel, R.; Nair, M.P.N.; Sufrin, G.; Mahajan, S.D.; Chadha, K.C.; Chawda, R.P.; Schwartz, S.A. Gene Expression of Angiogenic Factors Correlates with Metastatic Potential of Prostate Cancer Cells. Cancer Res. 2004, 64, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor Angiogenesis and Metastasis—Correlation in Invasive Breast Carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.W.C.; Poon, R.T.P. Clinical implications of angiogenesis in cancers. Vasc. Health Risk Manag. 2006, 2, 97–108. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, H.; Sakurai, S.; Liu, X.; Migita, H.; Wang, H.; Yamagishi, S.; Nomura, M.; Abedin, M.J.; Unoki, H.; Yamamoto, Y.; et al. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J. Biol. Chem. 1999, 274, 5172–5178. [Google Scholar] [CrossRef]
- Hauser, S.; Weich, H.A. A heparin-binding form of placenta growth factor (PlGF-2) is expressed in human umbilical vein endothelial cells and in placenta. Growth Factors 1993, 9, 259–268. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.-Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; et al. A Model of Human Tumor Dormancy: An Angiogenic Switch from the Nonangiogenic Phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef]
- Dunn, L.K.; Mohammad, K.S.; Fournier, P.G.J.; McKenna, C.R.; Davis, H.W.; Niewolna, M.; Peng, X.H.; Chirgwin, J.M.; Guise, T.A. Hypoxia and TGF-β Drive Breast Cancer Bone Metastases through Parallel Signaling Pathways in Tumor Cells and the Bone Microenvironment. PLoS ONE 2009, 4, e6896. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhau, H.E.; Huang, W.-C.; Iqbal, S.; Habib, F.K.; Sartor, O.; Cvitanovic, L.; Marshall, F.F.; Xu, Z.; Chung, L.W.K. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: Implication in human prostate cancer bone metastasis. Oncogene 2007, 26, 5070–5077. [Google Scholar] [CrossRef]
- Chim, S.M.; Tickner, J.; Chow, S.T.; Kuek, V.; Guo, B.; Zhang, G.; Rosen, V.; Erber, W.; Xu, J. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013, 24, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Trebec-Reynolds, D.P.; Voronov, I.; Heersche, J.N.; Manolson, M.F. VEGF-A expression in osteoclasts is regulated by NF-κB induction of HIF-1α. J. Cell. Biochem. 2010, 110, 343–351. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Zhou, Z.; Shi, H.; Qiu, X.; Xiong, J.; Chen, Y. BDNF regulates the expression and secretion of VEGF from osteoblasts via the TrkB/ERK1/2 signaling pathway during fracture healing. Mol. Med. Rep. 2017, 15, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tian, W.; Li, Y.; Tang, W.; Zhang, C. Osteoblast-specific transcription factor Osterix (Osx) and HIF-1α cooperatively regulate gene expression of vascular endothelial growth factor (VEGF). Biochem. Biophys. Res. Commun. 2012, 424, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular Endothelial Growth Factor and Angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Tombran-Tink, J.; Barnstable, C. Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: Possible mediators of angiogenesis and matrix remodeling in the bone. Biochem. Biophys. Res. Commun. 2004, 316, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Bruni-Cardoso, A.; Johnson, L.C.; Vessella, R.L.; Peterson, T.E.; Lynch, C.C. Osteoclast-Derived Matrix Metalloproteinase-9 Directly Affects Angiogenesis in the Prostate Tumor–Bone Microenvironment. Mol. Cancer Res. 2010, 8, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Nakano, A.; Takeuchi, K.; Kitazoe, K.; Kido, S.; Inoue, D.; et al. Myeloma Cell-Osteoclast Interaction Enhances Angiogenesis Together with Bone Resorption: A Role for Vascular Endothelial Cell Growth Factor and Osteopontin. Clin. Cancer Res. 2007, 13, 816–823. [Google Scholar] [CrossRef]
- Mulcrone, P.L.; Campbell, J.P.; Clément-Demange, L.; Anbinder, A.L.; Merkel, A.R.; Brekken, R.A.; Sterling, J.A.; Elefteriou, F. Skeletal Colonization by Breast Cancer Cells Is Stimulated by an Osteoblast and β2AR-Dependent Neo-Angiogenic Switch. J. Bone Miner. Res. 2017, 32, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Crawford, R.; Xiao, Y. The ratio of VEGF/PEDF expression in bone marrow mesenchymal stem cells regulates neovascularization. Differ. Res. Biol. Divers. 2011, 81, 181–191. [Google Scholar] [CrossRef]
- Akiyama, T.; Dass, C.R.; Shinoda, Y.; Kawano, H.; Tanaka, S.; Choong, P.F. PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem. Biophys. Res. Commun. 2010, 391, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Ek, E.T.H.; Dass, C.R.; Contreras, K.G.; Choong, P.F.M. PEDF-derived synthetic peptides exhibit antitumor activity in an orthotopic model of human osteosarcoma. J. Orthop. Res. 2007, 25, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, E.R.; Chen, L.; Fine, D.; Zhao, Y.; Mascarinas, E.; Grippo, P.J.; DiPietro, L.A. Pigment Epithelium-Derived Factor (PEDF) as a Regulator of Wound Angiogenesis. Sci. Rep. 2018, 8, 11142. [Google Scholar] [CrossRef]
- Yin, J.J.; Zhang, L.; Munasinghe, J.; Linnoila, R.I.; Kelly, K. Cediranib/AZD2171 Inhibits Bone and Brain Metastasis in a Preclinical Model of Advanced Prostate Cancer. Cancer Res. 2010, 70, 8662–8673. [Google Scholar] [CrossRef]
- Dahut, W.L.; Madan, R.A.; Karakunnel, J.J.; Adelberg, D.; Gulley, J.L.; Turkbey, I.B.; Chau, C.H.; Spencer, S.D.; Mulquin, M.; Wright, J.; et al. Phase II clinical trial of cediranib in patients with metastatic castration-resistant prostate cancer. BJU Int. 2013, 111, 1269–1280. [Google Scholar] [CrossRef]
- Heath, E.; Heilbrun, L.; Mannuel, H.; Liu, G.; Lara, P.; Monk, J.P.; Flaig, T.; Zurita, A.; Mack, P.; Vaishampayan, U.; et al. Phase II, Multicenter, Randomized Trial of Docetaxel plus Prednisone with or Without Cediranib in Men with Chemotherapy-Naive Metastatic Castrate-Resistant Prostate Cancer. Oncology 2019, 24, 1149-e807. [Google Scholar] [CrossRef] [PubMed]
- Bubeník, J. Tumour MHC class I downregulation and immunotherapy (Review). Oncol. Rep. 2003, 10, 2005–2008. [Google Scholar] [CrossRef] [PubMed]
- Baschuk, N.; Rautela, J.; Parker, B.S. Bone specific immunity and its impact on metastasis. BoneKEy Rep. 2015, 4, 665. [Google Scholar] [CrossRef]
- Akfirat, C.; Zhang, X.; Ventura, A.; Berel, D.; Colangelo, M.E.; Miranti, C.K.; Krajewska, M.; Reed, J.C.; Higano, C.S.; True, L.D.; et al. Tumour cell survival mechanisms in lethal metastatic prostate cancer differ between bone and soft tissue metastases. J. Pathol. 2013, 230, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Lickliter, J.D.; Cox, J.; McCarron, J.; Martinez, N.R.; Schmidt, C.W.; Lin, H.; Nieda, M.; Nicol, A.J. Small-molecule Bcl-2 inhibitors sensitise tumour cells to immune-mediated destruction. Br. J. Cancer 2007, 96, 600–608. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting Edge: TGF-β Induces a Regulatory Phenotype in CD4+CD25− T Cells through Foxp3 Induction and Down-Regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; Van Herwijnen, M.J.C.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef]
- Solis-Castillo, L.A.; Garcia-Romo, G.S.; Diaz-Rodriguez, A.; Reyes-Hernandez, D.; Tellez-Rivera, E.; Rosales-Garcia, V.H.; Mendez-Cruz, A.R.; Jimenez-Flores, J.R.; Villafana-Vazquez, V.H.; Pedroza-Gonzalez, A. Tumor-infiltrating regulatory T cells, CD8/Treg ratio, and cancer stem cells are correlated with lymph node metastasis in patients with early breast cancer. Breast Cancer 2020, 27, 837–849. [Google Scholar] [CrossRef]
- Marshall, E.A.; Ng, K.W.; Kung, S.H.Y.; Conway, E.M.; Martinez, V.D.; Halvorsen, E.C.; Rowbotham, D.A.; Vucic, E.A.; Plumb, A.W.; Becker-Santos, D.D.; et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol. Cancer 2016, 15, 1–15. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Zhao, E.; Wang, L.; Dai, J.; Kryczek, I.; Wei, S.; Vatan, L.; Altuwaijri, S.; Sparwasser, T.; Wang, G.; Keller, E.T.; et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. OncoImmunology 2012, 1, 152–161. [Google Scholar] [CrossRef]
- Zou, L.; Barnett, B.; Safah, H.; LaRussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone Marrow Is a Reservoir for CD4+CD25+ Regulatory T Cells that Traffic through CXCL12/CXCR4 Signals. Cancer Res. 2004, 64, 8451–8455. [Google Scholar] [CrossRef] [PubMed]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 axis—untapped potential in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 140, 50–66. [Google Scholar] [CrossRef]
- Zeelenberg, I.S.; Ruuls-Van Stalle, L.; Roos, E. The Chemokine Receptor CXCR4 Is Required for Outgrowth of Colon Carcinoma Micrometastases. Cancer Res. 2003, 63, 3833–3839. [Google Scholar]
- Huang, S.; Wei, P.; Hwang-Verslues, W.W.; Kuo, W.; Jeng, Y.; Hu, C.; Shew, J.; Huang, C.; Chang, K.; Lee, E.Y.; et al. TGF-β1 secreted by Tregs in lymph nodes promotes breast cancer malignancy via up-regulation of IL-17RB. EMBO Mol. Med. 2017, 9, 1660–1680. [Google Scholar] [CrossRef]
- Weiner, H.L. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 2001, 182, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Strober, W. Cell Contact–Dependent Immunosuppression by Cd4+Cd25+Regulatory T Cells Is Mediated by Cell Surface–Bound Transforming Growth Factor β. J. Exp. Med. 2001, 194, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; Von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF- signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Leal, A.C.; Gonçalves-Silva, T.; Mercadante, A.C.T.; Kestelman, F.; Chaves, S.B.; Azevedo, R.B.; Monteiro, J.P.; Bonomo, A. T Cells Induce Pre-Metastatic Osteolytic Disease and Help Bone Metastases Establishment in a Mouse Model of Metastatic Breast Cancer. PLoS ONE 2013, 8, e68171. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, D.; Shi, Q.; Huang, X.; Ju, X. Umbilical cord blood-derived Helios-positive regulatory T cells promote angiogenesis in acute lymphoblastic leukemia in mice via CCL22 and the VEGFA-VEGFR2 pathway. Mol. Med. Rep. 2019, 19, 4195–4204. [Google Scholar] [CrossRef]
- Zhan, H.-L.; Gao, X.; Zhou, X.-F.; Pu, X.-Y.; Wang, D.-J. Presence of Tumour-infiltrating FOXP3+Lymphocytes Correlates with Immature Tumour Angiogenesis in Renal Cell Carcinomas. Asian Pac. J. Cancer Prev. 2012, 13, 867–872. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Bates, G.J.; Koukourakis, M.I.; Sivridis, E.; Gatter, K.C.; Harris, A.L.; Banham, A.H. The presence of tumor-infiltrating FOXP3+ lymphocytes correlates with intratumoral angiogenesis in endometrial cancer. Gynecol. Oncol. 2008, 110, 216–221. [Google Scholar] [CrossRef]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and Interleukin-6 Promote Survival of Human CD11b+ Peripheral Blood Mononuclear Cells and Induce M2-type Macrophage Polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Carmeliet, P. Bridges that guide and unite. Nat. Cell Biol. 2010, 465, 697–699. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Pollard, J.W. Tumor-Associated Macrophages Press the Angiogenic Switch in Breast Cancer: Figure. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.-F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.-N.; Pollard, J.W. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Kaur, S.; Raggatt, L.J.; Batoon, L.; Hume, D.A.; Levesque, J.-P.; Pettit, A.R. Role of bone marrow macrophages in controlling homeostasis and repair in bone and bone marrow niches. Semin. Cell Dev. Biol. 2017, 61, 12–21. [Google Scholar] [CrossRef]
- Chang, M.K.; Raggatt, L.-J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal Tissue Macrophages Are Intercalated throughout Human and Mouse Bone Lining Tissues and Regulate Osteoblast Function In Vitro and In Vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef]
- Alexander, K.A.; Raggatt, L.-J.; Millard, S.; Batoon, L.; Wu, A.C.-K.; Chang, M.-K.; Hume, D.A.; Pettit, A.R. Resting and injury-induced inflamed periosteum contain multiple macrophage subsets that are located at sites of bone growth and regeneration. Immunol. Cell Biol. 2016, 95, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Soki, F.N.; Koh, A.J.; Eber, M.R.; Entezami, P.; Park, S.I.; Van Rooijen, N.; McCauley, L.K. Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc. Natl. Acad. Sci. USA 2014, 111, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Nicolaidou, V.; Wong, M.M.; Redpath, A.N.; Ersek, A.; Baban, D.F.; Williams, L.M.; Cope, A.P.; Horwood, N.J. Monocytes Induce STAT3 Activation in Human Mesenchymal Stem Cells to Promote Osteoblast Formation. PLoS ONE 2012, 7, e39871. [Google Scholar] [CrossRef] [PubMed]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Rédini, F.; Heymann, D.; et al. Induction of Osteogenesis in Mesenchymal Stem Cells by Activated Monocytes/Macrophages Depends on Oncostatin M Signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Pirraco, R.P.; Reis, R.L.; Marques, A.P. Effect of monocytes/macrophages on the early osteogenic differentiation of hBMSCs. J. Tissue Eng. Regen. Med. 2013, 7, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.J.; Hodge, J.M.; Singh, P.P.; Eeles, D.G.; Collier, F.M.; Holten, I.; Ebeling, P.R.; Nicholson, G.C.; Quinn, J.M.W. Cord Blood-Derived Macrophage-Lineage Cells Rapidly Stimulate Osteoblastic Maturation in Mesenchymal Stem Cells in a Glycoprotein-130 Dependent Manner. PLoS ONE 2013, 8, e73266. [Google Scholar] [CrossRef] [PubMed]
- Vi, L.; Baht, G.S.; Whetstone, H.; Qingxia, W.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages Promote Osteoblastic Differentiation In Vivo: Implications in Fracture Repair and Bone Homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef]
- Batoon, L.; Millard, S.M.; Raggatt, L.J.; Pettit, A.R. Osteomacs and Bone Regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Pettit, A.R.; Chang, M.K.; Hume, D.A.; Raggatt, L.-J. Osteal macrophages: A new twist on coupling during bone dynamics. Bone 2008, 43, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their Emerging Roles in Bone. J. Bone Miner. Res. 2015, 30, 2140–2149. [Google Scholar] [CrossRef]
- Soki, F.N.; Cho, S.W.; Kim, Y.W.; Jones, J.D.; Park, S.I.; Koh, A.J.; Entezami, P.; Daignault-Newton, S.; Pienta, K.J.; Roca, H.; et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015, 6, 35782–35796. [Google Scholar] [CrossRef]
- Wu, A.C.; He, Y.; Broomfield, A.; Paatan, N.J.; Harrington, B.S.; Tseng, H.-W.; Beaven, E.A.; Kiernan, D.M.; Swindle, P.; Clubb, A.B.; et al. CD169+macrophages mediate pathological formation of woven bone in skeletal lesions of prostate cancer. J. Pathol. 2016, 239, 218–230. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation Induces Myeloid-Derived Suppressor Cells that Facilitate Tumor Progression. J. Immunol. 2005, 176, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1β-Secreting Cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 Promotes Tumor Progression by Inducing Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A.; Xu, M.; Hadinoto, V.; Appanna, R.; et al. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.L.; Kumar, V.; Martner, A.; Mony, S.; Donthireddy, L.; Condamine, T.; Seykora, J.; Knight, S.C.; Malietzis, G.; Lee, G.H.; et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17–producing CD4+ T cells. J. Exp. Med. 2015, 212, 351–367. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-Derived Suppressor Cells Enhance Stemness of Cancer Cells by Inducing MicroRNA101 and Suppressing the Corepressor CtBP2. Immun. 2013, 39, 611–621. [Google Scholar] [CrossRef]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef]
- Sawant, A.; Deshane, J.; Jules, J.; Lee, C.M.; Harris, B.A.; Feng, X.; Ponnazhagan, S. Myeloid-Derived Suppressor Cells Function as Novel Osteoclast Progenitors Enhancing Bone Loss in Breast Cancer. Cancer Res. 2013, 73, 672–682. [Google Scholar] [CrossRef]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S.; et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef]
- Niu, G.; Bowman, T.; Huang, M.; Shivers, S.; Reintgen, D.; Daud, A.; Chang, A.; Kraker, A.; Jove, R.; Yu, H. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 2002, 21, 7001–7010. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Kremer, M.; Specht, K.; Calzada-Wack, J.; Nathrath, M.; Schaich, R.; Höfler, H.; Fend, F. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am. J. Pathol. 2003, 162, 1449–1461. [Google Scholar] [CrossRef]
- Diaz, N.; Minton, S.; Cox, C.; Bowman, T.; Gritsko, T.; Garcia, R.; Eweis, I.; Wloch, M.; Livingston, S.; Seijo, E.; et al. Activation of Stat3 in Primary Tumors from High-Risk Breast Cancer Patients Is Associated with Elevated Levels of Activated Src and Survivin Expression. Clin. Cancer Res. 2006, 12, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent Activation of Stat3 Signaling Induces Survivin Gene Expression and Confers Resistance to Apoptosis in Human Breast Cancer Cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Hamaguchi, T.; Nagao, N.; Kato, S.; Iino, T.; Nakamura, T.; Sudo, A. Interleukin-6 receptor inhibitor suppresses bone metastases in a breast cancer cell line. Breast Cancer 2018, 25, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, F.; Duplomb, L.; Baud’Huin, M.; Brounais, B. The dual role of IL-6-type cytokines on bone remodeling and bone tumors. Cytokine Growth Factor Rev. 2009, 20, 19–28. [Google Scholar] [CrossRef]
- Guillen, C.; De Gortázar, A.R.; Esbrit, P. The Interleukin-6/Soluble Interleukin-6 Receptor System Induces Parathyroid Hormone–Related Protein in Human Osteoblastic Cells. Calcif. Tissue Int. 2004, 75, 153–159. [Google Scholar] [CrossRef]
- Erices, A.; Congeta, P.; Rojasa, C.; Minguellab, J.J. gp130 Activation by Soluble Interleukin-6 Receptor/Interleukin-6 Enhances Osteoblastic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells. Exp. Cell Res. 2002, 280, 24–32. [Google Scholar] [CrossRef]
- Fan, Y.; Ye, J.; Shen, F.; Zhu, Y.; Yeghiazarians, Y.; Zhu, W.; Chen, Y.; Lawton, M.T.; Young, W.L.; Yang, G.-Y. Interleukin-6 Stimulates Circulating Blood-Derived Endothelial Progenitor Cell Angiogenesis in vitro. Br. J. Pharmacol. 2007, 28, 90–98. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Jee, S.-H.; Chu, C.-Y.; Chiu, H.-C.; Huang, Y.-L.; Tsai, W.-L.; Liao, Y.-H.; Kuo, M.-L. Interleukin-6 Induced Basic Fibroblast Growth Factor-Dependent Angiogenesis in Basal Cell Carcinoma Cell Line via JAK/STAT3 and PI3-Kinase/Akt Pathways. J. Investig. Dermatol. 2004, 123, 1169–1175. [Google Scholar] [CrossRef]
- Huang, S.-P.; Wu, M.-S.; Shun, C.-T.; Wang, H.-P.; Lin, M.-T.; Kuo, M.-L.; Lin, J.-T. Interleukin-6 increases vascular endothelial growth factor and angiogenesis in gastric carcinoma. J. Biomed. Sci. 2004, 11, 517–527. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Santegoets, S.J.A.M.; Reyners, A.K.L.; Goedemans, R.; Wouters, M.C.A.; Kenter, G.G.; Van Erkel, A.R.; Van Poelgeest, M.I.E.; Nijman, H.W.; Van Der Hoeven, J.J.M.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Decker, A.M.; Decker, J.T.; Jung, Y.; Cackowski, F.C.; Daignault-Newton, S.; Morgan, T.M.; Shea, L.D.; Taichman, R.S. Adrenergic Blockade Promotes Maintenance of Dormancy in Prostate Cancer Through Upregulation of GAS6. Transl. Oncol. 2020, 13, 100781. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.-S.; Shao, A.-W.; Ou, Y.-B.; Guo, Z.-N.; Manaenko, A.; Dixon, B.J.; Tang, J.; Lou, M.; Zhang, J.H. Recombinant Gas6 augments Axl and facilitates immune restoration in an intracerebral hemorrhage mouse model. Br. J. Pharmacol. 2016, 37, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-M.; Lee, E.-H.; Lee, H.-Y.; Choi, H.-R.; Ji, K.-Y.; Kim, S.-M.; Kim, K.D.; Kang, H.-S. Axl signaling induces development of natural killer cells in vitro and in vivo. Protoplasma 2016, 254, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Pardali, E.; Van Der Horst, G.; Cheung, H.; Hoogen, C.V.D.; Van Der Pluijm, G.; Dijke, P.T. Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene 2009, 29, 1351–1361. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Bajou, K.; Noel, A.; Gerard, R.D.; Masson, V.; Brunner, N.; Holsthansen, C.; Skobe, M.; Fusenig, N.E.; Carmeliet, P.; Collen, D.; et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat. Med. 1998, 4, 923–928. [Google Scholar] [CrossRef]
- Shimo, T.; Nakanishi, T.; Nishida, T.; Asano, M.; Kanyama, M.; Kuboki, T.; Tamatani, T.; Tezuka, K.; Takemura, M.; Matsumura, T.; et al. Connective Tissue Growth Factor Induces the Proliferation, Migration, and Tube Formation of Vascular Endothelial Cells In Vitro, and Angiogenesis In Vivo. J. Biochem. 1999, 126, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Ge, R.; Grazioli, A.; Xie, W.; Banach-Petrosky, W.; Kang, Y.; Lonning, S.; McPherson, J.; Yingling, J.M.; Biswas, S.; et al. Targeting the Transforming Growth Factor-β pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 2010, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.A.; Heier, A. Induction of Heart Valve Lesions by Small-Molecule ALK5 Inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Kang, J.-H.; Jung, M.-Y.; Yin, X.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-β signaling. J. Clin. Investig. 2017, 127, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Repellin, C.E.; Kang, J.-H.; Andrianifahanana, M.; Yin, X.; Leof, E.B. Sorting nexin 9 differentiates ligand-activated Smad3 from Smad2 for nuclear import and transforming growth factor β signaling. Mol. Biol. Cell 2015, 26, 3879–3891. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Kubota, S.; Yoshioka, N.; Ibaragi, S.; Isowa, S.; Eguchi, T.; Sasaki, A.; Takigawa, M. Pathogenic Role of Connective Tissue Growth Factor (CTGF/CCN2) in Osteolytic Metastasis of Breast Cancer. J. Bone Miner. Res. 2006, 21, 1045–1059. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Stevenson, J.P.; Kindler, H.L.; Papasavvas, E.; Sun, J.; Jacobs-Small, M.; Hull, J.; Schwed, D.; Ranganathan, A.; Newick, K.; Heitjan, D.F.; et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. OncoImmunology 2013, 2, e26218. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef]
- Bagi, C.M.; Roberts, G.W.; Andresen, C.J. Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors. Cancer 2008, 112, 2313–2321. [Google Scholar] [CrossRef]
- Kurio, N.; Shimo, T.; Fukazawa, T.; Takaoka, M.; Okui, T.; Hassan, N.M.M.; Honami, T.; Hatakeyama, S.; Ikeda, M.; Naomoto, Y.; et al. Anti-tumor effect in human breast cancer by TAE226, a dual inhibitor for FAK and IGF-IR in vitro and in vivo. Exp. Cell Res. 2011, 317, 1134–1146. [Google Scholar] [CrossRef]
- Hirt, U.A.; Waizenegger, I.C.; Schweifer, N.; Haslinger, C.; Gerlach, D.; Braunger, J.; Weyer-Czernilofsky, U.; Stadtmüller, H.; Sapountzis, I.; Bader, G.; et al. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Hu, C.; Chen, X.; Wen, J.; Gong, L.; Liu, Z.; Wang, J.; Liang, J.; Hu, F.; Zhou, Q.; Wei, L.; et al. Antitumor effect of focal adhesion kinase inhibitor PF562271 against human osteosarcomain vitroandin vivo. Cancer Sci. 2017, 108, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.J.; Thomas, K.; Huang, C.S.; Gutknecht, M.F.; Botchwey, E.A.; Bouton, A.H. Regulation of osteoclast structure and function by FAK family kinases. J. Leukoc. Biol. 2012, 92, 1021–1028. [Google Scholar] [CrossRef]
- Cabrita, M.A.; Jones, L.M.; Quizi, J.L.; Sabourin, L.A.; McKay, B.C.; Addison, C.L. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol. Oncol. 2011, 5, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; McGivern, N.; Canel, M.; Byron, A.; Johnson, S.C.; McSorley, H.J.; Quinn, N.; Taggart, D.; Von Kreigsheim, A.; Anderton, S.M.; et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal. 2017, 10, eaan8355. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; Von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef]
- De Jonge, M.J.A.; Steeghs, N.; Lolkema, M.P.; Hotte, S.J.; Hirte, H.W.; Van Der Biessen, D.A.J.; Razak, A.R.A.; De Vos, F.Y.F.L.; Verheijen, R.B.; Schnell, D.; et al. Phase I Study of BI 853520, an Inhibitor of Focal Adhesion Kinase, in Patients with Advanced or Metastatic Nonhematologic Malignancies. Target. Oncol. 2019, 14, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef]
- Macdonald, T.J.; Taga, T.; Shimada, H.; Tabrizi, P.; Zlokovic, B.V.; Cheresh, D.A.; Laug, W.E. Preferential Susceptibility of Brain Tumors to the Antiangiogenic Effects of an αv Integrin Antagonist. Neurosurgery 2001, 48, 151–157. [Google Scholar] [CrossRef]
- Buerkle, M.A.; Pahernik, S.A.; Sutter, A.; Jonczyk, A.; Messmer, K.; Dellian, M. Inhibition of the alpha-ν integrins with a cyclic RGD peptide impairs angiogenesis, growth and metastasis of solid tumours in vivo. Br. J. Cancer 2002, 86, 788–795. [Google Scholar] [CrossRef]
- Mitjans, F.; Meyer, T.; Fittschen, C.; Goodman, S.; Jonczyk, A.; Marshall, J.F.; Reyes, G.; Piulats, J. In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. International journal of cancer. J. Int. Cancer 2000, 87, 716–723. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation. Anti-Cancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Bretschi, M.; Cheng, C.; Witt, H.; Dimitrakopoulou-Strauss, A.; Strauss, L.G.; Semmler, W.; Bäuerle, T. Cilengitide affects tumor compartment, vascularization and microenvironment in experimental bone metastases as shown by longitudinal 18F-FDG PET and gene expression analysis. J. Cancer Res. Clin. Oncol. 2012, 139, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Chillà, A.; Bianconi, D.; Geetha, N.; Dorda, A.; Poettler, M.; Unseld, M.; Sykoutri, D.; Redlich, K.; Zielinski, C.C.; Prager, G.W. Effects of cilengitide in osteoclast maturation and behavior. Exp. Cell Res. 2015, 337, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-Methylated Cyclic RGD Peptides as Highly Active and Selective αVβ3Integrin Antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Bäuerle, T.; Komljenovic, D.; Merz, M.; Berger, M.R.; Goodman, S.L.; Semmler, W. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using VCT, MRI and DCE-MRI in a longitudinal in vivo study. Int. J. Cancer 2010, 128, 2453–2462. [Google Scholar] [CrossRef]
- Bretschi, M.; Merz, M.; Komljenovic, D.; Berger, M.R.; Semmler, W.; Bäuerle, T. Cilengitide inhibits metastatic bone colonization in a nude rat model. Oncol. Rep. 2011, 26, 843–851. [Google Scholar] [CrossRef]
- Besse, B.; Tsao, L.C.; Chao, D.T.; Fang, Y.; Soria, J.-C.; Almokadem, S.; Belani, C.P. Phase Ib safety and pharmacokinetic study of volociximab, an anti-α5β1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann. Oncol. 2012, 24, 90–96. [Google Scholar] [CrossRef]
- Hussain, M.; Le Moulec, S.; Gimmi, C.; Bruns, R.; Straub, J.; Miller, K. Differential Effect on Bone Lesions of Targeting Integrins: Randomized Phase II Trial of Abituzumab in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 3192–3200. [Google Scholar] [CrossRef]
- Bishop, R.T.; Marino, S.; De Ridder, D.; Allen, R.J.; Lefley, D.V.; Sims, A.H.; Wang, N.; Ottewell, P.D.; Idris, A.I. Pharmacological inhibition of the IKKε/TBK-1 axis potentiates the anti-tumour and anti-metastatic effects of Docetaxel in mouse models of breast cancer. Cancer Lett. 2019, 450, 76–87. [Google Scholar] [CrossRef]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.L.; Zheng, Y.; Lam, H.-M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 39, 4421–4435. [Google Scholar] [CrossRef]
- Nobutani, K.; Shimono, Y.; Mizutani, K.; Ueda, Y.; Suzuki, T.; Kitayama, M.; Minami, A.; Momose, K.; Miyawaki, K.; Akashi, K.; et al. Downregulation of CXCR4 in Metastasized Breast Cancer Cells and Implication in Their Dormancy. PLoS ONE 2015, 10, e0130032. [Google Scholar] [CrossRef] [PubMed]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pang, Y.; Xie, T.; Zhu, L. CXCR4 antagonism in combination with IDO1 inhibition weakens immune suppression and inhibits tumor growth in mouse breast cancer bone metastases. OncoTargets Ther. 2019, 12, 4985–4992. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal Localization and Neutralization of the SDF-1(CXCL12)/CXCR4 Axis Blocks Prostate Cancer Metastasis and Growth in Osseous Sites In Vivo. J. Bone Miner. Res. 2004, 20, 318–329. [Google Scholar] [CrossRef]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar] [CrossRef]
- Litchfield, L.M.; Klinge, C.M. Multiple roles of COUP-TFII in cancer initiation and progression. J. Mol. Endocrinol. 2012, 49, R135–R148. [Google Scholar] [CrossRef]
- Borgen, E.; Rypdal, M.C.; Sosa, M.S.; Renolen, A.; Schlichting, E.; Lønning, P.E.; Synnestvedt, M.; Aguirre-Ghiso, J.A.; Naume, B. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 2018, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Decker, A.M.; Jung, Y.; Cackowski, F.C.; Yumoto, K.; Wang, J.; Taichman, R.S. Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow. Mol. Cancer Res. 2017, 15, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.-J.; Zheng, J.-Y.; Bian, J.-L.; Chen, L.-W.; Dong, N.; Yu, Y.; Hong, G.-L.; Chandoo, A.; Yao, Y.-M.; Lu, Z.-Q. Growth Arrest-Specific 6 Enhances the Suppressive Function of CD4+CD25+Regulatory T Cells Mainly through Axl Receptor. Mediat. Inflamm. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Hölzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; McDermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 Controls the Response to Multiple Cancer Drugs through Regulation of TGF-β Receptor Signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.G.; Juárez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-β Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef]
- Wan, X.; Li, Z.-G.; Yingling, J.M.; Yang, J.; Starbuck, M.W.; Ravoori, M.K.; Kundra, V.; Vazquez, E.; Navone, N.M. Effect of transforming growth factor beta (TGF-β) receptor I kinase inhibitor on prostate cancer bone growth. Bone 2012, 50, 695–703. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef]
- Kelley, R.; Gane, E.; Assenat, E.; Siebler, J.; Galle, P.; Merle, P.; Hourmand, I.; Cleverly, A.; Zhao, Y.; Gueorguieva, I.; et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2019, 10, e00056. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.-J.; Redd, R.A.; Perez, R.P.; Baz, R.; Zavidij, O.; Sklavenitis-Pistofidis, R.; Richardson, P.G.; Anderson, K.C.; Laubach, J.P.; et al. A Phase Ib/II Trial of the First-in-Class Anti-CXCR4 Antibody Ulocuplumab in Combination with Lenalidomide or Bortezomib Plus Dexamethasone in Relapsed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 344–353. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Liu, C.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Park, S.-Y.; Nam, J.-S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.H.; Esser, A.K.; Fox, G.C.; Schmieder, A.H.; Yang, X.; Hu, G.; Pan, D.; Su, X.; Xu, Y.; Novack, D.V.; et al. Bone-Induced Expression of Integrin β3 Enables Targeted Nanotherapy of Breast Cancer Metastases. Cancer Res. 2017, 77, 6299–6312. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; DeClerck, Y.A. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Harousseau, J.-L.; Wijdenes, J.; Morineau, N.; Milpied, N.; Bataille, R. A combination of anti-interleukin 6 murine monoclonal antibody with dexamethasone and high-dose melphalan induces high complete response rates in advanced multiple myeloma. Br. J. Haematol. 2000, 109, 661–664. [Google Scholar] [CrossRef]
- Dorff, T.B.; Goldman, B.; Pinski, J.K.; Mack, P.C.; Lara, P.N.; Van Veldhuizen, P.J.; Quinn, D.I.; Vogelzang, N.J.; Thompson, I.M.; Hussain, M.H. Clinical and Correlative Results of SWOG S0354: A Phase II Trial of CNTO328 (Siltuximab), a Monoclonal Antibody against Interleukin-6, in Chemotherapy-Pretreated Patients with Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2010, 16, 3028–3034. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; De Bono, J.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.; Qi, M.; Bandekar, R.; Vermeulen, J.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting Signal-Transducer-and-Activator-of-Transcription-3 for Prevention and Therapy of Cancer: Modern Target but Ancient Solution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169. [Google Scholar] [CrossRef]
- Gu, L.; Talati, P.; Vogiatzi, P.; Romero-Weaver, A.L.; Abdulghani, J.; Liao, Z.; Leiby, B.; Hoang, D.T.; Mirtti, T.; Alanen, K.; et al. Pharmacologic Suppression of JAK1/2 by JAK1/2 Inhibitor AZD1480 Potently Inhibits IL-6–Induced Experimental Prostate Cancer Metastases Formation. Mol. Cancer Ther. 2014, 13, 1246–1258. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and Antimetastatic Activity of JAK Inhibitor AZD1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef]
- Plimack, E.R.; Lorusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. Oncology 2013, 18, 819–820. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hutzen, B.; Zuo, M.; Ball, S.; DeAngelis, S.; Foust, E.; Pandit, B.; Ihnat, M.A.; Shenoy, S.S.; Kulp, S.; et al. Novel STAT3 Phosphorylation Inhibitors Exhibit Potent Growth-Suppressive Activity in Pancreatic and Breast Cancer Cells. Cancer Res. 2010, 70, 2445–2454. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]


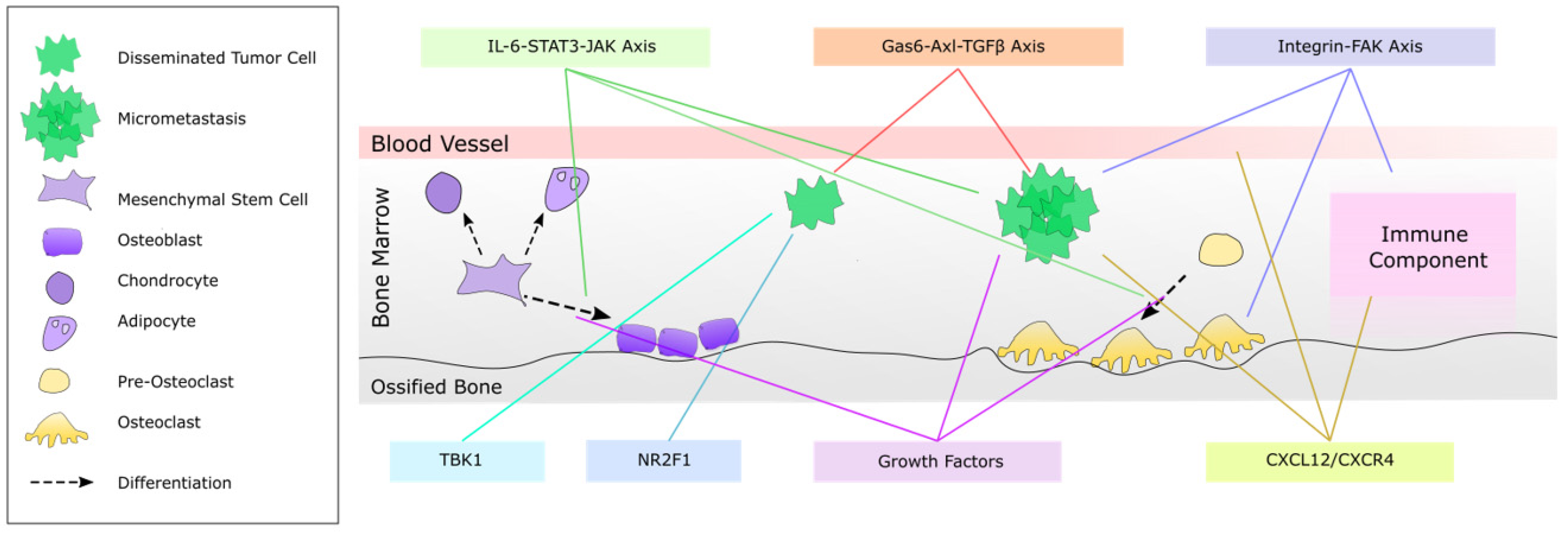{kind=link}
| Pathway | Factor (s) | Effect | Potential Therapy |
|---|---|---|---|
| IL-6-STAT3-JAK Axis | IL-6 | Tumor cell survival [241,242,243,244,245] Tumor cell expression of osteogenic and angiogenic factors, VEGF and RANK [246] Tumor cell and osteoblast production of RANKL and PTHrP to induce osteoclastogenesis [247,248,249] Angiogenesis [250,251,252,253] | IL-6R antagonists [254,246] Jagged1 inhibitor, 15D11, which decreases osteoblast IL-6 production [129] |
| GAS6-Axl-TGF-β Axis | GAS6 expression by osteoblasts | Activation of Axl, which is required for TGFβ2-mediated tumor cell dormancy [61] | Propranolol [255] Recombinant GAS6 [256] Agonistic antibodies to Axl [257] |
| TGFβR1/2, SMAD3 or downstream CTGF and PAI-1 | Tumor cell growth [258] Tumor cell expression of PTHrP [259] Angiogenesis [260,261] | TGFβR1 inhibitor LY2109761 [262] or LY2157299 [263] Specific peptide antagonists of SMAD3 [260,261,264,265,266] TGF-β antibody, fresolimumab [267,268,269] | |
| Integrin-FAK Axis | FAK | Bone metastatic growth and osteolysis [270,271,272,273] Expression of osteogenic and angiogenic factor, RANKL [271] Osteoclast differentiation and function [274] Angiogenesis [275] Recruitment of Tregs [276,277] | FAK inhibitors [270,271,272,273,274,275,276,277,278,279,280] |
| Integrin α5β3, α5β5, α5β1 | Expression of osteogenic and angiogenic factors, RANKL, VEGF and PTHrP [281,282,283,284,285] Osteoclast maturation and function [286] | Cilengitide [281,282,283,284,285,286,287,288,289] Integrin α5β1 antibody antagonists [290] Abituzumab [291] | |
| TBK1 | Stromal cell induction of tumor cell TBK1expression | Chemoresistance via inhibition of mTOR [81] | TBK1 inhibitor, Amlexanox [292] |
| NR2F1 | NR2F1 expression by tumor cells | Tumor cell dormancy [293] | Activators of tumor cell NR2F1, such as combination 5-Aza-C and retinoic acid, to induce or maintain tumor cell dormancy [293] |
| Growth Factors | VEGF isoforms by osteoblasts, osteoclasts and tumor cells, and P1GF by vasculature | Tumor cell adhesion [153] Angiogenesis [153,155] Osteoblastogenesis [134,135,136] Osteolytic metastasis [140,141] | VEGF inhibitor, cediranib [170,171,172] PEDF-derived peptides [167] or recombinant PEDF [168,169] to suppress angiogenesis, bone resorption and VEGF expression |
| CXCL12-CXCR4 | CXCL12-CXCR4 interaction | Tumor cell proliferation [49,294,295] Metastatic growth [296,297,298] Tumor mass angiogenesis [299] Intratumoral infiltration of MDSC and Tregs [297] Systemic CD8+ and CD4+ T cells [297] | Plerixafor [296] AMD3465 [297] TC14012 [298] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreps, L.M.; Addison, C.L. Targeting Intercellular Communication in the Bone Microenvironment to Prevent Disseminated Tumor Cell Escape from Dormancy and Bone Metastatic Tumor Growth. Int. J. Mol. Sci. 2021, 22, 2911. https://doi.org/10.3390/ijms22062911
Kreps LM, Addison CL. Targeting Intercellular Communication in the Bone Microenvironment to Prevent Disseminated Tumor Cell Escape from Dormancy and Bone Metastatic Tumor Growth. International Journal of Molecular Sciences. 2021; 22(6):2911. https://doi.org/10.3390/ijms22062911
Chicago/Turabian StyleKreps, Lauren M., and Christina L. Addison. 2021. "Targeting Intercellular Communication in the Bone Microenvironment to Prevent Disseminated Tumor Cell Escape from Dormancy and Bone Metastatic Tumor Growth" International Journal of Molecular Sciences 22, no. 6: 2911. https://doi.org/10.3390/ijms22062911
APA StyleKreps, L. M., & Addison, C. L. (2021). Targeting Intercellular Communication in the Bone Microenvironment to Prevent Disseminated Tumor Cell Escape from Dormancy and Bone Metastatic Tumor Growth. International Journal of Molecular Sciences, 22(6), 2911. https://doi.org/10.3390/ijms22062911