
Physical Exercise and Alzheimer’s Disease: Effects on Pathophysiological Molecular Pathways of the Disease



 ,
,  , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Physical Activity and Alzheimer’s Disease
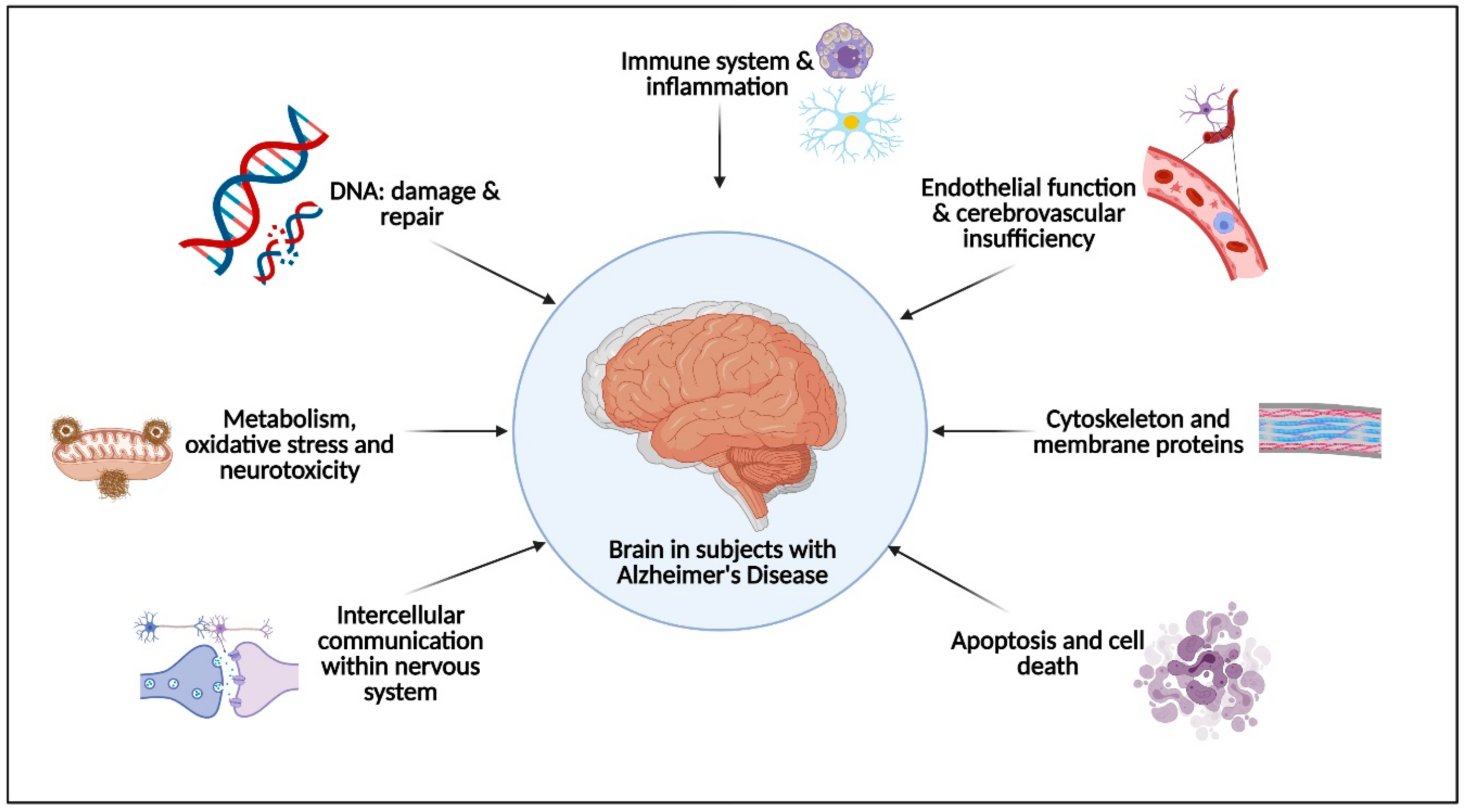
2. Physical Activity Effects on the Pathophysiological Molecular Pathways Associated with Alzheimer’s Disease
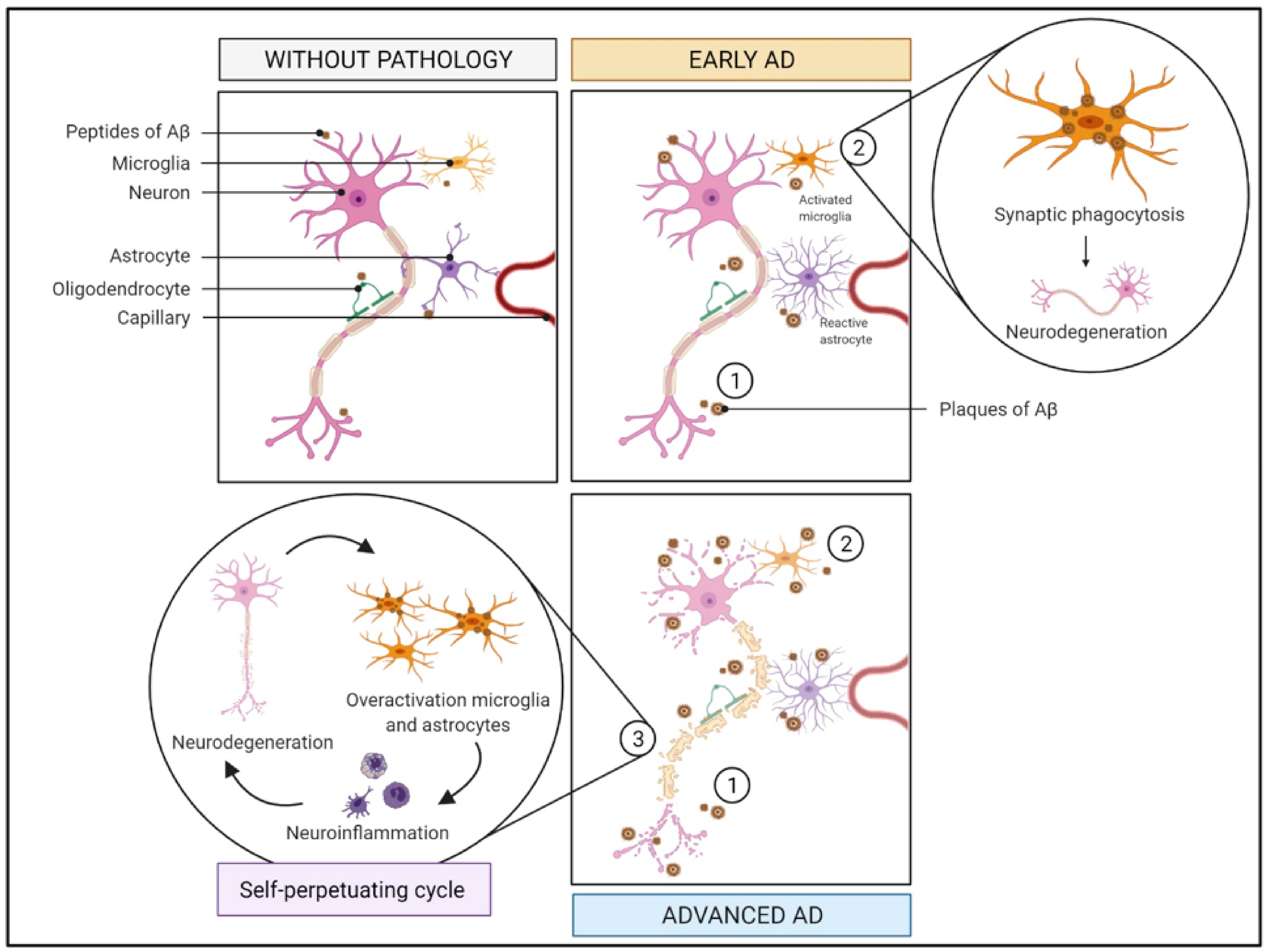
2.1. Immune System and Inflammation
2.1.1. What Is It? How Does It Relate to AD?
2.1.2. How Does It Relate to Physical Activity?
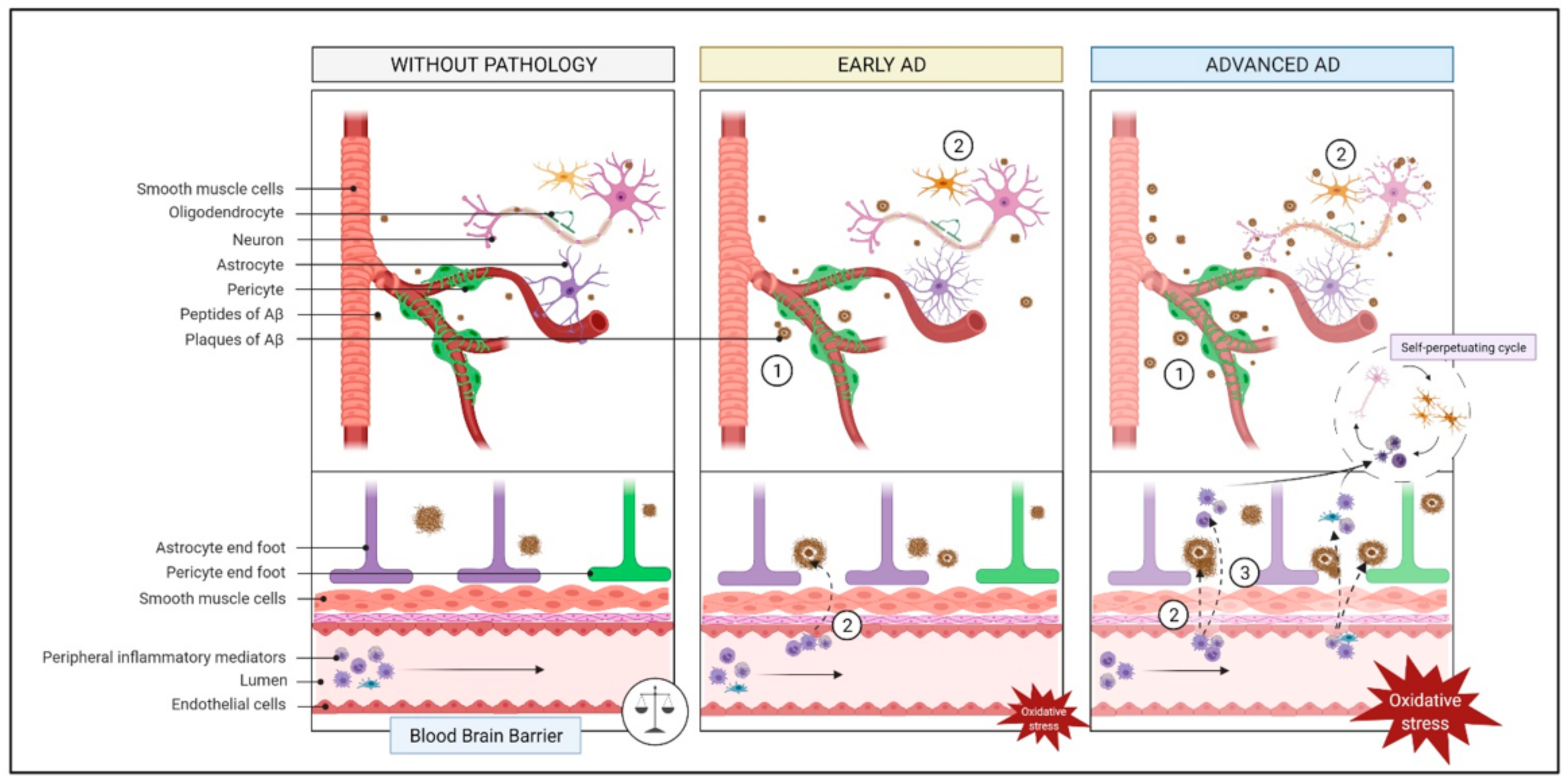
2.2. Endothelial Function and Cerebrovascular Insufficiency
2.2.1. What Is It? How Does It Relate to AD?
2.2.2. How Does It Relate to Physical Activity?
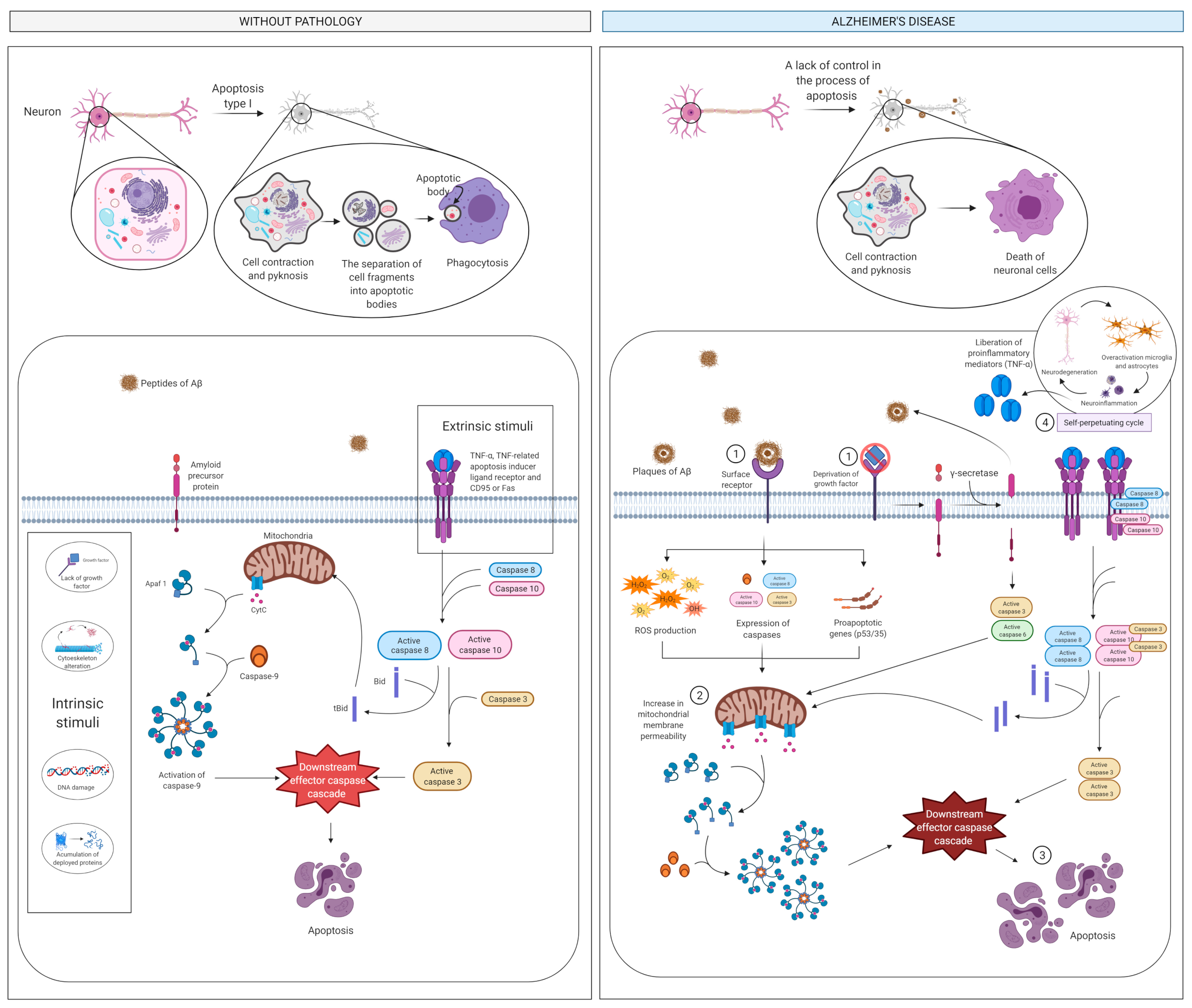
2.3. Apoptosis and Cell Death
2.3.1. What Is It? How Does It Relate to AD?
2.3.2. How Does It Relate to Physical Activity?
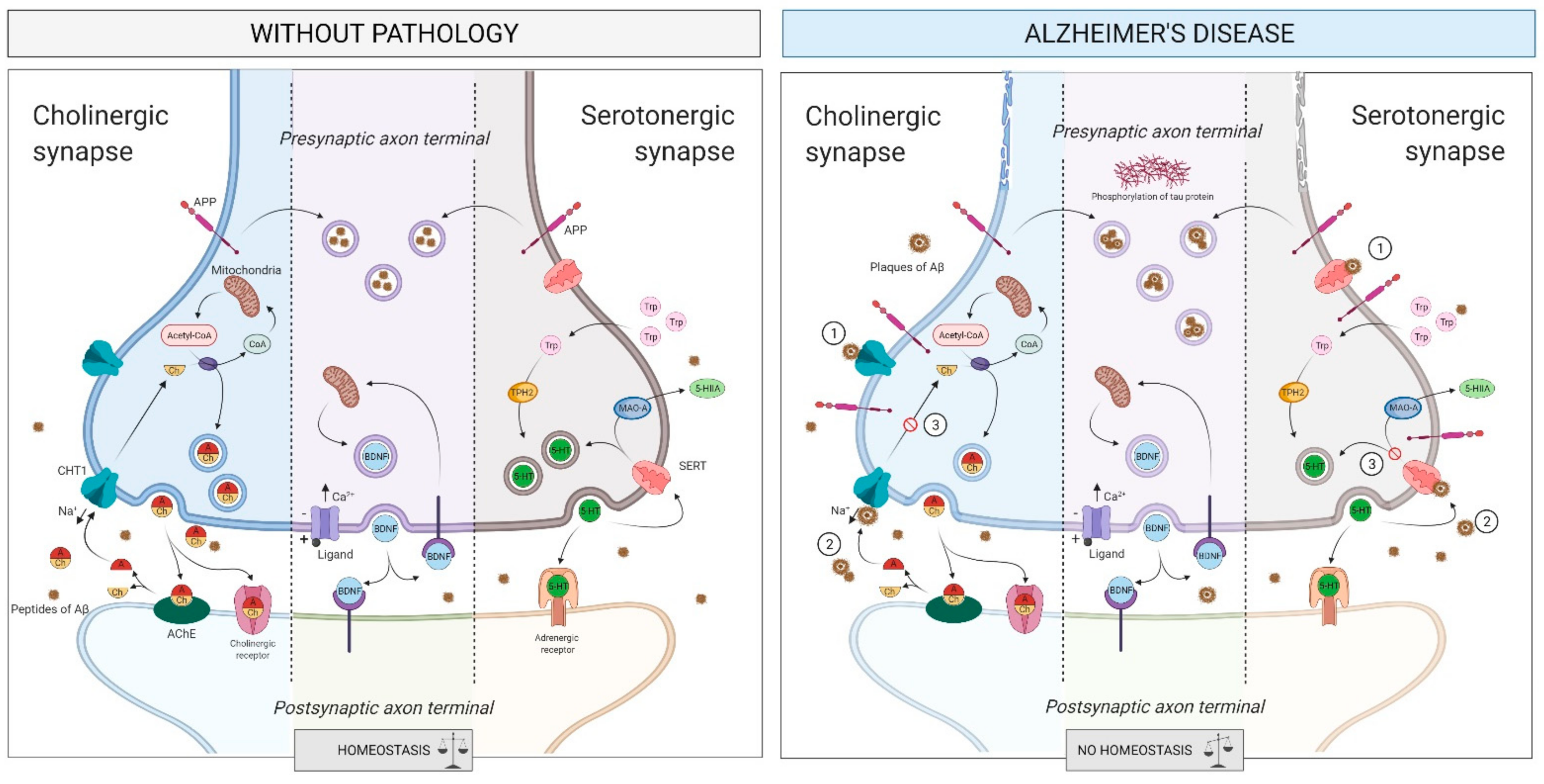
2.4. Intercellular Communication
2.4.1. What Is It? How Does It Relate to AD?
The Cholinergic System
The Serotonergic System
2.4.2. How Does It Relate to Physical Activity?
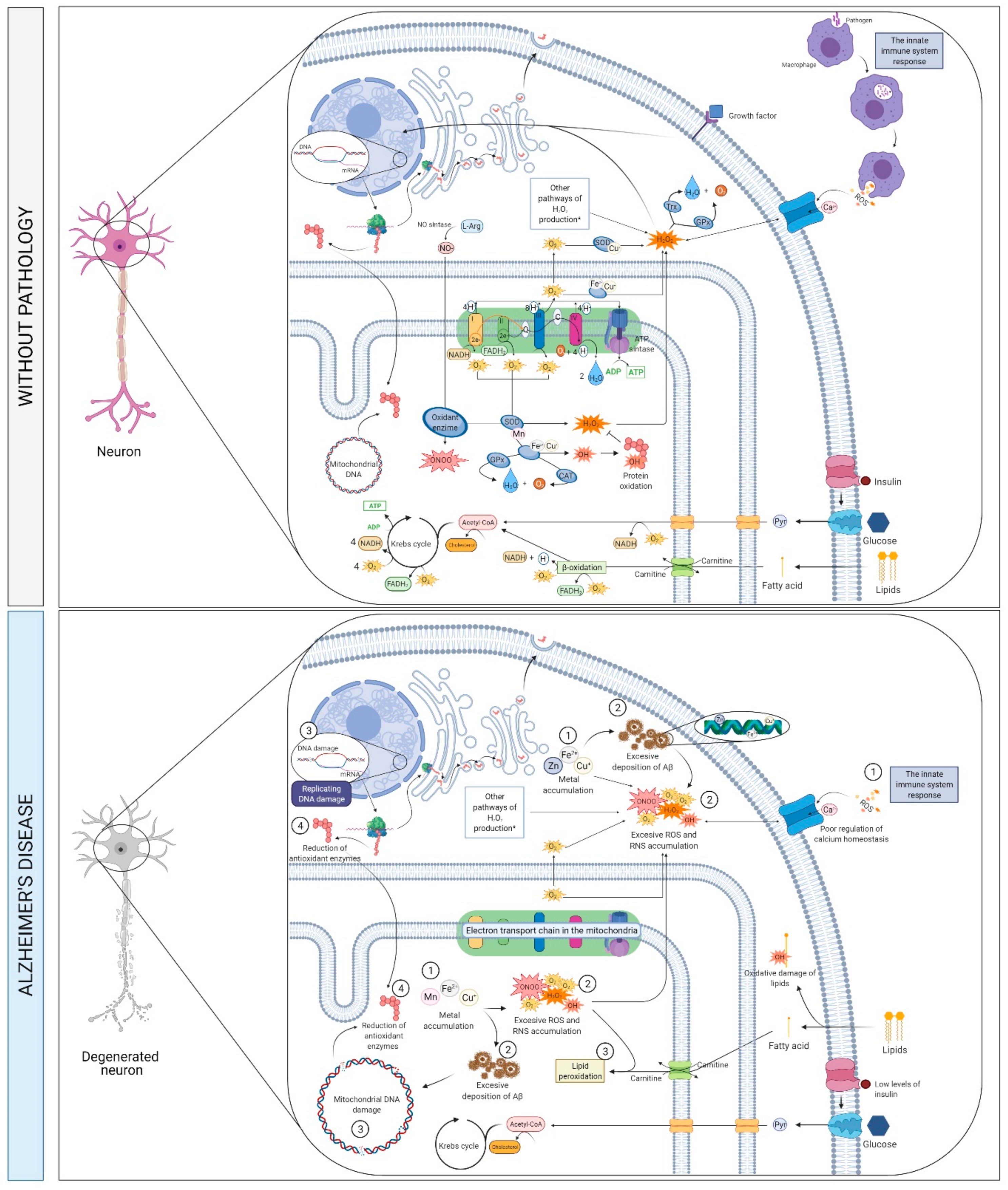
2.5. Metabolims, Oxidative Stress and Neurotoxicity
2.5.1. What Is It? How Does It Relate to AD?
APP Proteolytic Processing
2.5.2. How Does It Relate to Physical Activity?
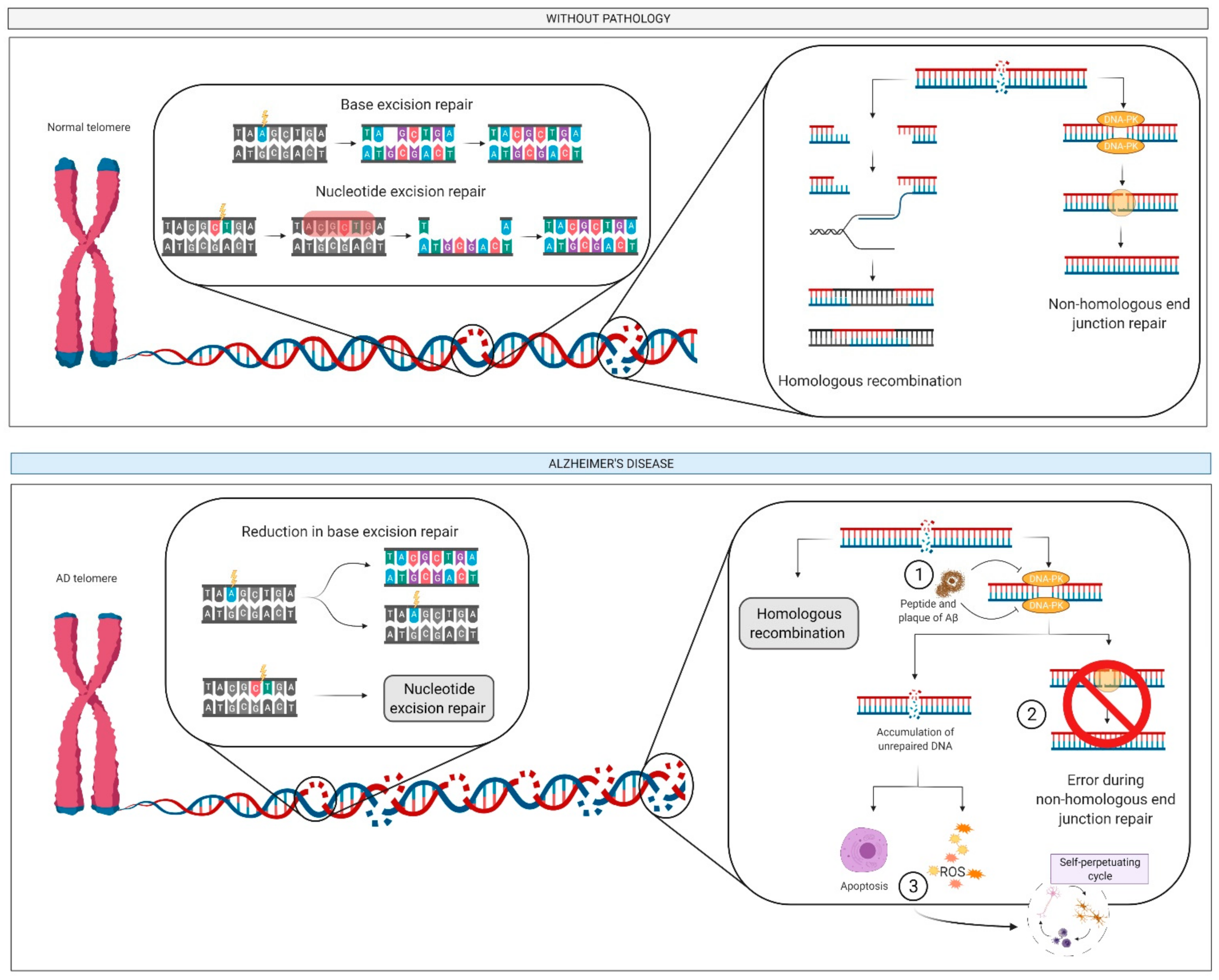
2.6. DNA: Damage and Repair
2.6.1. What Is It? How Does it Relate to AD?
2.6.2. How Does It Relate to Physical Activity?
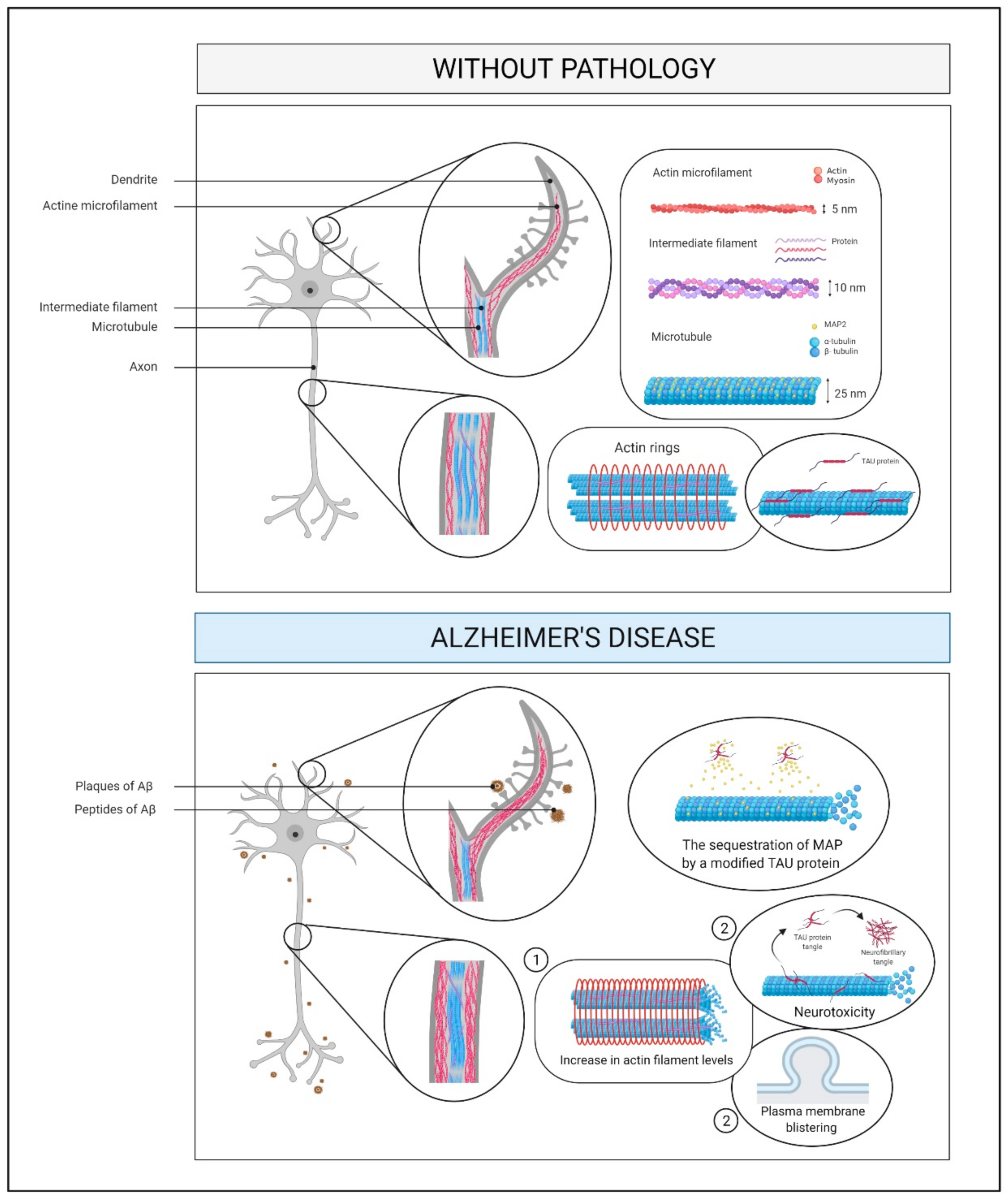
2.7. Cytoskeleton and Membrane Proteins
2.7.1. What Is It? How Does It Relate to AD?
2.7.2. How Does It Relate to Physical Activity?
2.8. Synaptic Plasticity
2.8.1. What Is It? How Does It Relate to AD?
2.8.2. How Does It Relate to Physical Activity?
3. Investigating the Link between Alzheimer’s Disease and Physical Exercise through Neuroimaging Technologies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Ageing 2017; United Nations Publications: New York, NY, USA, 2017. [Google Scholar]
- Macpherson, H.; Teo, W.P.; Schneider, L.A.; Smith, A.E. A Life-Long Approach to Physical Activity for Brain Health. Front. Aging Neurosci. 2017, 9, 147. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia an Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 10 February 2021).
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Castrillo, J.I.; Lista, S.; Hampel, H.; Ritchie, C.W. Systems Biology Methods for Alzheimer’s Disease Research Toward Molecular Signatures, Subtypes, and Stages and Precision Medicine: Application in Cohort Studies and Trials. Methods Mol. Biol. 2018, 1750, 31–66. [Google Scholar]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Koffie, R.M.; Hyman, B.T.; Spires-Jones, T.L. Alzheimer’s disease: Synapses gone cold. Mol. Neurodegener. 2011, 6, 63. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2018 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-onset Alzheimer Disease and Its Variants. Continuum 2019, 25, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Gustafson, D.R.; Hardy, J. The genetic architecture of Alzheimer’s disease: Beyond APP, PSENs and APOE. Neurobiol. Aging 2012, 33, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Jarmolowicz, A.I.; Chen, H.Y.; Panegyres, P.K. The patterns of inheritance in early-onset dementia: Alzheimer’s disease and frontotemporal dementia. Am. J. Alzheimers Dis. Other Demen. 2015, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of physical exercise in Alzheimer’s disease. Biomed. Rep. 2016, 4, 403–407. [Google Scholar] [CrossRef]
- Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Exercise as a Polypill for Chronic Diseases. Prog. Mol. Biol. Transl. Sci. 2015, 135, 497–526. [Google Scholar]
- Santos-Lozano, A.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Quindos-Rubial, M.; Fiuza-Luces, C.; Cristi-Montero, C.; Emanuele, E.; Garatachea, N.; Lucia, A. Physical Activity and Alzheimer Disease: A Protective Association. Mayo Clin. Proc. 2016, 91, 999–1020. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Garatachea, N.; Berger, N.A.; Lucia, A. Exercise is the real polypill. Physiology 2013, 28, 330–358. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Du, Z.; Li, Y.; Li, J.; Zhou, C.; Li, F.; Yang, X. Physical activity can improve cognition in patients with Alzheimer’s disease: A systematic review and meta-analysis of randomized controlled trials. Clin. Interv. Aging 2018, 13, 1593–1603. [Google Scholar] [CrossRef]
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181. [Google Scholar] [CrossRef]
- Rao, A.K.; Chou, A.; Bursley, B.; Smulofsky, J.; Jezequel, J. Systematic review of the effects of exercise on activities of daily living in people with Alzheimer’s disease. Am. J. Occup. Ther. 2014, 68, 50–56. [Google Scholar] [CrossRef]
- Zhu, X.C.; Yu, Y.; Wang, H.F.; Jiang, T.; Cao, L.; Wang, C.; Wang, J.; Tan, C.C.; Meng, X.F.; Tan, L.; et al. Physiotherapy intervention in Alzheimer’s disease: Systematic review and meta-analysis. J. Alzheimers Dis. 2015, 44, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, J.I.; Oliver, S.G. Alzheimer’s as a Systems-Level Disease Involving the Interplay of Multiple Cellular Networks. Methods Mol. Biol. 2016, 1303, 3–48. [Google Scholar]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Dammer, E.B.; Swarup, V.; Nandakumar, D.; Duong, D.M.; Yin, L.; Deng, Q.; Nguyen, T.; Hales, C.M.; Wingo, T.; et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017, 4, 60–72. [Google Scholar] [CrossRef]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Serpente, M.; Bonsi, R.; Scarpini, E.; Galimberti, D. Innate immune system and inflammation in Alzheimer’s disease: From pathogenesis to treatment. Neuroimmunomodulation 2014, 21, 79–87. [Google Scholar] [CrossRef]
- Jensen, C.J.; Massie, A.; De Keyser, J. Immune players in the CNS: The astrocyte. J. Neuroimmune Pharmacol. 2013, 8, 824–839. [Google Scholar] [CrossRef]
- Jevtic, S.; Sengar, A.S.; Salter, M.W.; McLaurin, J. The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res. Rev. 2017, 40, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.; Fuentes, M.; Alarcon, M.; Palomo, I. Immune System Dysfunction in the Elderly. An. Acad. Bras. Cienc. 2017, 89, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.J.; Quinlan, M.A.; Blakely, R.D. Immune System Activation and Depression: Roles of Serotonin in the Central Nervous System and Periphery. ACS Chem. Neurosci. 2017, 8, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Van Eldik, L.J.; Carrillo, M.C.; Cole, P.E.; Feuerbach, D.; Greenberg, B.D.; Hendrix, J.A.; Kennedy, M.; Kozauer, N.; Margolin, R.A.; Molinuevo, J.L.; et al. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimers Dement. 2016, 2, 99–109. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.V.; Town, T. Innate immunity in Alzheimer’s disease: A complex affair. CNS Neurol. Disord. Drug Targets 2013, 12, 593–607. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Physical activity and exercise attenuate neuroinflammation in neurological diseases. Brain Res. Bull. 2016, 125, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. Modifiable risk factors of Alzheimer’sDisease and neuroinflammation: What are the links? Future Neurol. 2016, 11, 237–244. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Brolinson, P.G.; Elliott, D. Exercise and the immune system. Clin. Sports Med. 2007, 26, 311–319. [Google Scholar] [CrossRef]
- Simpson, R.J.; Kunz, H.; Agha, N.; Graff, R. Exercise and the Regulation of Immune Functions. Prog. Mol. Biol. Transl. Sci. 2015, 135, 355–380. [Google Scholar]
- Mee-Inta, O.; Zhao, Z.W.; Kuo, Y.M. Physical Exercise Inhibits Inflammation and Microglial Activation. Cells 2019, 8, 691. [Google Scholar] [CrossRef]
- Vecchio, L.M.; Meng, Y.; Xhima, K.; Lipsman, N.; Hamani, C.; Aubert, I. The Neuroprotective Effects of Exercise: Maintaining a Healthy Brain Throughout Aging. Brain Plast. 2018, 4, 17–52. [Google Scholar] [CrossRef] [PubMed]
- Stephen, R.; Hongisto, K.; Solomon, A.; Lonnroos, E. Physical Activity and Alzheimer’s Disease: A Systematic Review. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2017, 72, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Soiza, R.L. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197–226. [Google Scholar]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Malkiewicz, M.A.; Szarmach, A.; Sabisz, A.; Cubala, W.J.; Szurowska, E.; Winklewski, P.J. Blood-brain barrier permeability and physical exercise. J. Neuroinflamm. 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Antunes, B.M.; Rossi, F.E.; Cholewa, J.M.; Lira, F.S. Regular Physical Activity and Vascular Aging. Curr. Pharm. Des. 2016, 22, 3715–3729. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Santos-Lozano, A.; Joyner, M.; Carrera-Bastos, P.; Picazo, O.; Zugaza, J.L.; Izquierdo, M.; Ruilope, L.M.; Lucia, A. Exercise benefits in cardiovascular disease: Beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 2018, 15, 731–743. [Google Scholar] [CrossRef]
- De Biase, C.; De Rosa, R.; Luciano, R.; De Luca, S.; Capuano, E.; Trimarco, B.; Galasso, G. Effects of physical activity on endothelial progenitor cells (EPCs). Front. Physiol. 2013, 4, 414. [Google Scholar] [CrossRef]
- Di Francescomarino, S.; Sciartilli, A.; Di Valerio, V.; Di Baldassarre, A.; Gallina, S. The effect of physical exercise on endothelial function. Sports Med. 2009, 39, 797–812. [Google Scholar] [CrossRef]
- Van der Kleij, L.A.; Petersen, E.T.; Siebner, H.R.; Hendrikse, J.; Frederiksen, K.S.; Sobol, N.A.; Hasselbalch, S.G.; Garde, E. The effect of physical exercise on cerebral blood flow in Alzheimer’s disease. Neuroimage Clin. 2018, 20, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Alkadhi, K.A. Exercise as a Positive Modulator of Brain Function. Mol. Neurobiol. 2018, 55, 3112–3130. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, T.; Chu, J.M.; Chen, Y.; Dunnett, S.; Ho, Y.S.; Wong, G.T.; Chang, R.C. The beneficial effects of physical exercise in the brain and related pathophysiological mechanisms in neurodegenerative diseases. Lab. Investig. 2019, 99, 943–957. [Google Scholar] [CrossRef]
- Sofi, F.; Valecchi, D.; Bacci, D.; Abbate, R.; Gensini, G.F.; Casini, A.; Macchi, C. Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. J. Intern. Med. 2011, 269, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. 3), S125–S152. [Google Scholar] [CrossRef]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Mooren, F.C.; Kruger, K. Exercise, Autophagy, and Apoptosis. Prog. Mol. Biol. Transl. Sci. 2015, 135, 407–422. [Google Scholar] [PubMed]
- Garatachea, N.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Santos-Lozano, A.; Fiuza-Luces, C.; Moran, M.; Emanuele, E.; Joyner, M.J.; Lucia, A. Exercise attenuates the major hallmarks of aging. Rejuvenation Res. 2015, 18, 57–89. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Pickett, E.; Spires-Jones, T.L. Synaptic pathology: A shared mechanism in neurological disease. Ageing Res. Rev. 2016, 28, 72–84. [Google Scholar] [CrossRef]
- Marcello, E.; Epis, R.; Saraceno, C.; Di Luca, M. Synaptic dysfunction in Alzheimer’s disease. Adv. Exp. Med. Biol. 2012, 970, 573–601. [Google Scholar]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M. The cholinergic lesion of Alzheimer’s disease: Pivotal factor or side show? Learn. Mem. 2004, 11, 43–49. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Mesulam, M.M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ginsberg, S.D.; Ikonomovic, M.D.; DeKosky, S.T. Human cholinergic basal forebrain: Chemoanatomy and neurologic dysfunction. J. Chem. Neuroanat. 2003, 26, 233–242. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M.M. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 9 (Suppl. 2), 23–28. [Google Scholar] [CrossRef]
- Mesulam, M. A horseradish peroxidase method for the identification of the efferents of acetyl cholinesterase-containing neurons. J. Histochem. Cytochem. 1976, 24, 1281–1285. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Arendt, T.; Taubert, G.; Bigl, V.; Arendt, A. Amyloid deposition in the nucleus basalis of Meynert complex: A topographic marker for degenerating cell clusters in Alzheimer’s disease. Acta Neuropathol. 1988, 75, 226–232. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Gil-Bea, F.J.; Garcia-Alloza, M.; Dominguez, J.; Marcos, B.; Ramirez, M.J. Evaluation of cholinergic markers in Alzheimer’s disease and in a model of cholinergic deficit. Neurosci. Lett. 2005, 375, 37–41. [Google Scholar] [CrossRef]
- Perry, E.K.; Blessed, G.; Tomlinson, B.E.; Perry, R.H.; Crow, T.J.; Cross, A.J.; Dockray, G.J.; Dimaline, R.; Arregui, A. Neurochemical activities in human temporal lobe related to aging and Alzheimer-type changes. Neurobiol. Aging 1981, 2, 251–256. [Google Scholar] [CrossRef]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 1978, 2, 1457–1459. [Google Scholar] [CrossRef]
- Baskin, D.S.; Browning, J.L.; Pirozzolo, F.J.; Korporaal, S.; Baskin, J.A.; Appel, S.H. Brain choline acetyltransferase and mental function in Alzheimer disease. Arch. Neurol. 1999, 56, 1121–1123. [Google Scholar] [CrossRef]
- Pappas, B.A.; Bayley, P.J.; Bui, B.K.; Hansen, L.A.; Thal, L.J. Choline acetyltransferase activity and cognitive domain scores of Alzheimer’s patients. Neurobiol. Aging 2000, 21, 11–17. [Google Scholar] [CrossRef]
- Meneses, A.; Perez-Garcia, G.; Ponce-Lopez, T.; Tellez, R.; Castillo, C. Serotonin transporter and memory. Neuropharmacology 2011, 61, 355–363. [Google Scholar] [CrossRef]
- Wirth, A.; Holst, K.; Ponimaskin, E. How serotonin receptors regulate morphogenic signalling in neurons. Prog. Neurobiol. 2017, 151, 35–56. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Fakhfouri, G.; Ramezani, V.; Mehr, S.E.; Rahimian, R. The role of serotonin in memory: Interactions with neurotransmitters and downstream signaling. Exp. Brain Res. 2014, 232, 723–738. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Noristani, H.N.; Verkhratsky, A. The serotonergic system in ageing and Alzheimer’s disease. Prog. Neurobiol. 2012, 99, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Wojsiat, J.; Laskowska-Kaszub, K.; Mietelska-Porowska, A.; Wojda, U. Search for Alzheimer’s disease biomarkers in blood cells: Hypotheses-driven approach. Biomark. Med. 2017, 11, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Shimodaira, Y.; Nakamura, H.; Imaizumi, A.; Mori, M.; Kageyama, Y.; Noguchi, Y.; Seki, A.; Okabe, Y.; Miyake, Y.; et al. Low plasma tryptophan is associated with olfactory function in healthy elderly community dwellers in Japan. BMC Geriatr. 2017, 17, 1–6. [Google Scholar] [CrossRef]
- Giil, L.M.; Midttun, O.; Refsum, H.; Ulvik, A.; Advani, R.; Smith, A.D.; Ueland, P.M. Kynurenine Pathway Metabolites in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 495–504. [Google Scholar] [CrossRef]
- Pena-Bautista, C.; Flor, L.; Lopez-Nogueroles, M.; Garcia, L.; Ferrer, I.; Baquero, M.; Vento, M.; Chafer-Pericas, C. Plasma alterations in cholinergic and serotonergic systems in early Alzheimer Disease: Diagnosis utility. Clin. Chim. Acta 2020, 500, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.V.; Proza, J.F.; Sohrabji, F.; Lawler, J.M. Vascular and metabolic dysfunction in Alzheimer’s disease: A review. Exp. Biol. Med. 2011, 236, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.; Grant, M.M.; Aldred, S. Oxidative stress in vascular dementia and Alzheimer’s disease: A common pathology. J. Alzheimers Dis. 2009, 17, 245–257. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Birla, H.; Minocha, T.; Kumar, G.; Misra, A.; Singh, S.K. Role of Oxidative Stress and Metal Toxicity in the Progression of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimers Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, G.; Song, J. The association between PGC-1alpha and Alzheimer’s disease. Anat. Cell Biol. 2016, 49, 1–6. [Google Scholar] [CrossRef]
- De Oliveira Bristot, V.J.; de Bem Alves, A.C.; Cardoso, L.R.; da Luz Scheffer, D.; Aguiar, A.S.J. The Role of PGC-1alpha/UCP2 Signaling in the Beneficial Effects of Physical Exercise on the Brain. Front. Neurosci. 2019, 13, 292. [Google Scholar] [CrossRef]
- Katsouri, L.; Parr, C.; Bogdanovic, N.; Willem, M.; Sastre, M. PPARgamma co-activator-1alpha (PGC-1alpha) reduces amyloid-beta generation through a PPARgamma-dependent mechanism. J. Alzheimers Dis. 2011, 25, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Panes, J.D.; Godoy, P.A.; Silva-Grecchi, T.; Celis, M.T.; Ramirez-Molina, O.; Gavilan, J.; Munoz-Montecino, C.; Castro, P.A.; Moraga-Cid, G.; Yevenes, G.E.; et al. Changes in PGC-1alpha/SIRT1 Signaling Impact on Mitochondrial Homeostasis in Amyloid-Beta Peptide Toxicity Model. Front. Pharmacol. 2020, 11, 709. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. The role of metabolic disorders in Alzheimer disease and vascular dementia: Two roads converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Bostrom, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J.; Begg, D.; Mathai, M.; Weisinger, R.S. Omega 3 fatty acids and the brain: Review of studies in depression. Asia Pac. J. Clin. Nutr. 2007, 16 (Suppl. 1), 391–397. [Google Scholar]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance—A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Agranoff, B.W.; Radin, N.S.; Burton, R.M. Comparison of the fatty acids of lipids of subcellular brain fractions. J. Neurochem. 1969, 16, 397–404. [Google Scholar] [CrossRef]
- Janssen, C.I.; Kiliaan, A.J. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: The influence of LCPUFA on neural development, aging, and neurodegeneration. Prog. Lipid Res. 2014, 53, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fonteh, A.N.; Cipolla, M.; Chiang, J.; Arakaki, X.; Harrington, M.G. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS ONE 2014, 9, e100519. [Google Scholar] [CrossRef]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimers Dement. 2017, 13, 810–827. [Google Scholar] [CrossRef]
- El Gaamouch, F.; Jing, P.; Xia, J.; Cai, D. Alzheimer’s Disease Risk Genes and Lipid Regulators. J. Alzheimers Dis. 2016, 53, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lu, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Tacal, O. Trafficking and proteolytic processing of amyloid precursor protein and secretases in Alzheimer’s disease development: An up-to-date review. Eur. J. Pharmacol. 2019, 856, 172415. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef]
- Grimm, M.O.; Haupenthal, V.J.; Mett, J.; Stahlmann, C.P.; Blumel, T.; Mylonas, N.T.; Endres, K.; Grimm, H.S.; Hartmann, T. Oxidized Docosahexaenoic Acid Species and Lipid Peroxidation Products Increase Amyloidogenic Amyloid Precursor Protein Processing. Neurodegener. Dis 2016, 16, 44–54. [Google Scholar] [CrossRef]
- Jean-Louis, T.; Rockwell, P.; Figueiredo-Pereira, M.E. Prostaglandin J2 promotes O-GlcNAcylation raising APP processing by alpha- and beta-secretases: Relevance to Alzheimer’s disease. Neurobiol. Aging 2018, 62, 130–145. [Google Scholar] [CrossRef]
- Mesa-Herrera, F.; Taoro-Gonzalez, L.; Valdes-Baizabal, C.; Diaz, M.; Marin, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Dart, C. Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.; Fabelo, N.; Fernandez-Echevarria, C.; Canerina-Amaro, A.; Rodriguez-Barreto, D.; Quinto-Alemany, D.; Mesa-Herrera, F.; Diaz, M. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 2016, 13, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Fabelo, N.; Martin, V.; Ferrer, I.; Gomez, T.; Marin, R. Biophysical alterations in lipid rafts from human cerebral cortex associate with increased BACE1/AbetaPP interaction in early stages of Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 1185–1198. [Google Scholar] [CrossRef]
- Giordano, V.; Peluso, G.; Iannuccelli, M.; Benatti, P.; Nicolai, R.; Calvani, M. Systemic and brain metabolic dysfunction as a new paradigm for approaching Alzheimer’s dementia. Neurochem. Res. 2007, 32, 555–567. [Google Scholar] [CrossRef]
- Sun, J.H.; Yu, J.T.; Tan, L. The role of cholesterol metabolism in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 947–965. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Linares, C.; Kurti, A.; Knight, J.A.; Heckman, M.G.; Diehl, N.N.; Shinohara, M.; Martens, Y.A.; et al. APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat. Commun. 2018, 9, 4388. [Google Scholar] [CrossRef]
- Jackson, P.A.; Pialoux, V.; Corbett, D.; Drogos, L.; Erickson, K.I.; Eskes, G.A.; Poulin, M.J. Promoting brain health through exercise and diet in older adults: A physiological perspective. J. Physiol. 2016, 594, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Young, M.F.; Valaris, S.; Wrann, C.D. A role for FNDC5/Irisin in the beneficial effects of exercise on the brain and in neurodegenerative diseases. Prog. Cardiovasc. Dis. 2019, 62, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Sumsuzzman, D.M.; Choi, J.; Kang, H.; Lee, S.R.; Hong, Y. Molecular and Functional Interaction of the Myokine Irisin with Physical Exercise and Alzheimer’s Disease. Molecules 2018, 23, 3229. [Google Scholar] [CrossRef]
- Lira, V.A.; Benton, C.R.; Yan, Z.; Bonen, A. PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E145–E161. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Beedie, C.; Jimenez, A. Differential effects of aerobic exercise, resistance training and combined exercise modalities on cholesterol and the lipid profile: Review, synthesis and recommendations. Sports Med. 2014, 44, 211–221. [Google Scholar] [CrossRef]
- Bosma, M. Lipid homeostasis in exercise. Drug Discov. Today 2014, 19, 1019–1023. [Google Scholar] [CrossRef]
- Muscella, A.; Stefano, E.; Lunetti, P.; Capobianco, L.; Marsigliante, S. The Regulation of Fat Metabolism During Aerobic Exercise. Biomolecules 2020, 10, 1699. [Google Scholar] [CrossRef]
- Simioni, C.; Zauli, G.; Martelli, A.M.; Vitale, M.; Sacchetti, G.; Gonelli, A.; Neri, L.M. Oxidative stress: Role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 2018, 9, 17181–17198. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Bergman, B.C.; Goodpaster, B.H. Exercise and Muscle Lipid Content, Composition, and Localization: Influence on Muscle Insulin Sensitivity. Diabetes 2020, 69, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Liu, Z. Vascular function, insulin action, and exercise: An intricate interplay. Trends Endocrinol. Metab. 2015, 26, 297–304. [Google Scholar] [CrossRef]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Danese, E.; Lippi, G.; Sanchis-Gomar, F.; Brocco, G.; Rizzo, M.; Banach, M.; Montagnana, M. Physical Exercise and DNA Injury: Good or Evil? Adv. Clin. Chem. 2017, 81, 193–230. [Google Scholar]
- Schmidt, R.H.; Nickerson, J.M.; Boatright, J.H. Exercise as Gene Therapy: BDNF and DNA Damage Repair. Asia Pac. J. Ophthalmol. 2016, 5, 309–311. [Google Scholar] [CrossRef]
- Ramaekers, F.C.; Bosman, F.T. The cytoskeleton and disease. J. Pathol. 2004, 204, 351–354. [Google Scholar] [CrossRef]
- Cabrales Fontela, Y.; Kadavath, H.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat. Commun. 2017, 8, 1981. [Google Scholar] [CrossRef]
- Brandt, R.; Bakota, L. Microtubule dynamics and the neurodegenerative triad of Alzheimer’s disease: The hidden connection. J. Neurochem. 2017, 143, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Bliss, T.V.; Collingridge, G.L.; Morris, R.G. Synaptic plasticity in health and disease: Introduction and overview. Philos Trans. R. Soc. Lond B Biol. Sci. 2014, 369, 20130129. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The synaptic Abeta hypothesis of Alzheimer disease. Nat. Neurosci. 2005, 8, 977–979. [Google Scholar] [CrossRef]
- Mango, D.; Saidi, A.; Cisale, G.Y.; Feligioni, M.; Corbo, M.; Nistico, R. Targeting Synaptic Plasticity in Experimental Models of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 778. [Google Scholar] [CrossRef]
- Nistico, R.; Pignatelli, M.; Piccinin, S.; Mercuri, N.B.; Collingridge, G. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol. Neurobiol. 2012, 46, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Tsai, S.F.; Kuo, Y.M. Physical Exercise Enhances Neuroplasticity and Delays Alzheimer’s Disease. Brain Plast. 2018, 4, 95–110. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Lopez-Ramos, J.C.; Gimenez-Llort, L.; Revilla, S.; Guerra, R.; Gruart, A.; Laferla, F.M.; Cristofol, R.; Delgado-Garcia, J.M.; Sanfeliu, C. Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J. Alzheimers Dis. 2011, 24, 421–454. [Google Scholar] [CrossRef]
- Liu, H.L.; Zhao, G.; Cai, K.; Zhao, H.H.; Shi, L.D. Treadmill exercise prevents decline in spatial learning and memory in APP/PS1 transgenic mice through improvement of hippocampal long-term potentiation. Behav. Brain Res. 2011, 218, 308–314. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, H.L.; Zhang, H.; Tong, X.J. Treadmill exercise enhances synaptic plasticity, but does not alter beta-amyloid deposition in hippocampi of aged APP/PS1 transgenic mice. Neuroscience 2015, 298, 357–366. [Google Scholar] [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Alkadhi, K.A. Moderate Treadmill Exercise Protects Synaptic Plasticity of the Dentate Gyrus and Related Signaling Cascade in a Rat Model of Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Salim, S.; Eriksen, J.L.; Alkadhi, K.A. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr. Alzheimer Res. 2013, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Alkadhi, K.A. Comparison of the Effect of Exercise on Late-Phase LTP of the Dentate Gyrus and CA1 of Alzheimer’s Disease Model. Mol. Neurobiol. 2016, 53, 6859–6868. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chai, G.S.; Wang, Z.H.; Hu, Y.; Li, X.G.; Ma, Z.W.; Wang, Q.; Wang, J.Z.; Liu, G.P. CaMKII-dependent dendrite ramification and spine generation promote spatial training-induced memory improvement in a rat model of sporadic Alzheimer’sdisease. Neurobiol. Aging 2015, 36, 867–876. [Google Scholar] [CrossRef]
- Jiang, X.; Chai, G.S.; Wang, Z.H.; Hu, Y.; Li, X.G.; Ma, Z.W.; Wang, Q.; Wang, J.Z.; Liu, G.P. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci. Rep. 2015, 5, 9488. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Goncalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Frederiksen, K.S.; Gjerum, L.; Waldemar, G.; Hasselbalch, S.G. Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. J. Alzheimers Dis. 2018, 61, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Tustison, N.J.; Cook, P.A.; Klein, A.; Song, G.; Das, S.R.; Duda, J.T.; Kandel, B.M.; van Strien, N.; Stone, J.R.; Gee, J.C.; et al. Large-scale evaluation of ANTs and FreeSurfer cortical thickness measurements. Neuroimage 2014, 99, 166–179. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef]
- Wagner, G.; Herbsleb, M.; de la Cruz, F.; Schumann, A.; Brunner, F.; Schachtzabel, C.; Gussew, A.; Puta, C.; Smesny, S.; Gabriel, H.W.; et al. Hippocampal structure, metabolism, and inflammatory response after a 6-week intense aerobic exercise in healthy young adults: A controlled trial. J. Cereb. Blood Flow Metab. 2015, 35, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, E.D.; Honea, R.A.; Billinger, S.A.; Swerdlow, R.H.; Burns, J.M. Cardiorespiratory fitness is associated with atrophy in Alzheimer’s and aging over 2 years. Neurobiol. Aging 2012, 33, 1624–1632. [Google Scholar] [CrossRef]
- Fellgiebel, A.; Yakushev, I. Diffusion tensor imaging of the hippocampus in MCI and early Alzheimer’s disease. J. Alzheimers Dis. 2011, 26 (Suppl. 3), 257–262. [Google Scholar] [CrossRef]
- Zhang, Y.; Schuff, N.; Jahng, G.H.; Bayne, W.; Mori, S.; Schad, L.; Mueller, S.; Du, A.T.; Kramer, J.H.; Yaffe, K.; et al. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease. Neurology 2007, 68, 13–19. [Google Scholar] [CrossRef]
- Douaud, G.; Jbabdi, S.; Behrens, T.E.; Menke, R.A.; Gass, A.; Monsch, A.U.; Rao, A.; Whitcher, B.; Kindlmann, G.; Matthews, P.M.; et al. DTI measures in crossing-fibre areas: Increased diffusion anisotropy reveals early white matter alteration in MCI and mild Alzheimer’s disease. Neuroimage 2011, 55, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.M.; Guadagni, V.; Mazerolle, E.L.; Hill, M.; Hogan, D.B.; Pike, G.B.; Poulin, M.J. Effect of aerobic exercise on white matter microstructure in the aging brain. Behav. Brain Res. 2019, 373, 112042. [Google Scholar] [CrossRef]
- Perea, R.D.; Vidoni, E.D.; Morris, J.K.; Graves, R.S.; Burns, J.M.; Honea, R.A. Cardiorespiratory fitness and white matter integrity in Alzheimer’s disease. Brain Imaging Behav. 2016, 10, 660–668. [Google Scholar] [CrossRef]
- Van Osch, M.J.P.; Lu, H. Arterial Spin Labeling Perfusion MRI in Alzheimer’s Disease. Curr. Med. Imaging 2011, 7, 62–72. [Google Scholar] [CrossRef]
- Austin, B.P.; Nair, V.A.; Meier, T.B.; Xu, G.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Prabhakaran, V. Effects of hypoperfusion in Alzheimer’s disease. J. Alzheimers Dis. 2011, 26 (Suppl. 3), 123–133. [Google Scholar] [CrossRef]
- Colcombe, S.J.; Kramer, A.F.; Erickson, K.I.; Scalf, P.; McAuley, E.; Cohen, N.J.; Webb, A.; Jerome, G.J.; Marquez, D.X.; Elavsky, S. Cardiovascular fitness, cortical plasticity, and aging. Proc. Natl. Acad. Sci. USA 2004, 101, 3316–3321. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.W.; Prakash, R.S.; Erickson, K.I.; Basak, C.; Chaddock, L.; Kim, J.S.; Alves, H.; Heo, S.; Szabo, A.N.; White, S.M.; et al. Plasticity of brain networks in a randomized intervention trial of exercise training in older adults. Front. Aging Neurosci. 2010, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Spasov, S.; Passamonti, L.; Duggento, A.; Lio, P.; Toschi, N. Alzheimer’s Disease Neuroimaging, I. A parameter-efficient deep learning approach to predict conversion from mild cognitive impairment to Alzheimer’s disease. Neuroimage 2019, 189, 276–287. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Ortiz, S.; Pinto-Fraga, J.; Valenzuela, P.L.; Martín-Hernández, J.; Seisdedos, M.M.; García-López, O.; Toschi, N.; Di Giuliano, F.; Garaci, F.; Mercuri, N.B.; et al. Physical Exercise and Alzheimer’s Disease: Effects on Pathophysiological Molecular Pathways of the Disease. Int. J. Mol. Sci. 2021, 22, 2897. https://doi.org/10.3390/ijms22062897
López-Ortiz S, Pinto-Fraga J, Valenzuela PL, Martín-Hernández J, Seisdedos MM, García-López O, Toschi N, Di Giuliano F, Garaci F, Mercuri NB, et al. Physical Exercise and Alzheimer’s Disease: Effects on Pathophysiological Molecular Pathways of the Disease. International Journal of Molecular Sciences. 2021; 22(6):2897. https://doi.org/10.3390/ijms22062897
Chicago/Turabian StyleLópez-Ortiz, Susana, Jose Pinto-Fraga, Pedro L. Valenzuela, Juan Martín-Hernández, María M. Seisdedos, Oscar García-López, Nicola Toschi, Francesca Di Giuliano, Francesco Garaci, Nicola Biagio Mercuri, and et al. 2021. "Physical Exercise and Alzheimer’s Disease: Effects on Pathophysiological Molecular Pathways of the Disease" International Journal of Molecular Sciences 22, no. 6: 2897. https://doi.org/10.3390/ijms22062897
APA StyleLópez-Ortiz, S., Pinto-Fraga, J., Valenzuela, P. L., Martín-Hernández, J., Seisdedos, M. M., García-López, O., Toschi, N., Di Giuliano, F., Garaci, F., Mercuri, N. B., Nisticò, R., Emanuele, E., Lista, S., Lucia, A., & Santos-Lozano, A. (2021). Physical Exercise and Alzheimer’s Disease: Effects on Pathophysiological Molecular Pathways of the Disease. International Journal of Molecular Sciences, 22(6), 2897. https://doi.org/10.3390/ijms22062897