
Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape?


 , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
IPF: Pathobiologic Traits Recalling Malignant Proliferation
2. Cancer and IPF: Genetic, Epigenetic, Signaling Links and Differential Traits
2.1. Genetic Alterations
2.1.1. Oncogenes
2.1.2. Tumor Suppressor Genes
2.2. Microsatellites and Telomeres
2.3. Epigenetic Determinants
2.4. Biologic Divergences
3. Stemness-Like Networks
4. Common Available Biomarkers
5. Is There a Role for Immune Checkpoints in IPF?
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Coultas, D.B. The epidemioogy of idiopathic pulmonary fibrosis. In Seminar in Respiratory and Critical Care Medicine; Thieme Medical Publishers: Stuttgart, Germany, 1993; pp. 181–196. [Google Scholar]
- Coultas, D.B.; Zumwalt, R.E.; Black, W.C.; Sobonya, R.E. The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1994, 150, 967–972. [Google Scholar] [CrossRef]
- Meltzer, E.B.; Noble, P.W. Idiopathic pulmonary fibrosis. Orphanet J. Rare Dis. 2008, 3, 1–15. [Google Scholar] [CrossRef]
- Kim, D.S. Classification and Natural history of the idiopathic interstitial pneumonias. Proc. Am. Thorac. Soc. 2006, 3, 285–292. [Google Scholar] [CrossRef]
- Cameli, P.; Refini, R.M.; Bergantini, L.; D’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-term follow-up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: A real-life comparison study. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Panos, R.J.; Mortenson, R.L.; Niccoli, S.; King, T.E. Clinical deterioration in patients with idiopathic pulmonary fibrosis: Causes and assessment. Am. J. Med. 1990, 88, 396–404. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.; Mannino, D.M. Pulmonary fibrosis and lung cancer in the United States: Analysis of the multiple cause of death mortality data, 1979 through 1991. South. Med. J. 1996, 89, 505–510. [Google Scholar] [CrossRef]
- Artinian, V.; Kvale, P.A. Cancer and interstitial lung disease. Curr. Opin. Pulm. Med. 2004, 10, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic pulmonary fibrosis and lung cancer: Mechanisms and molecular targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Ehlers-Tenenbaum, S.; Schaaf, M.; Oltmanns, U.; Palmowski, K.; Hoffmann, H.; Schnabel, P.A.; Heußel, C.-P.; Puderbach, M.; Herth, F.J.F.; et al. Treatment and outcome of lung cancer in idiopathic interstitial pneumonias. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 31, 266–274. [Google Scholar]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Aubry, M.-C.; Myers, J.L.; Douglas, W.W.; Tazelaar, H.D.; Stephens, T.L.W.; Hartman, T.E.; Deschamps, C.; Pankratz, V.S. Primary pulmonary carcinoma in patients with idiopathic pulmonary fibrosis. Mayo Clin. Proc. 2002, 77, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef]
- Kato, E.; Takayanagi, N.; Takaku, Y.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Sugita, Y. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2018, 4, 00111–02016. [Google Scholar] [CrossRef]
- Nagai, A.; Chiyotani, A.; Nakadate, T.; Konno, K. Lung cancer in patients with idiopathic pulmonary fibrosis. Tohoku J. Exp. Med. 1992, 167, 231–237. [Google Scholar] [CrossRef]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef]
- Vancheri, C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur. Respir. Rev. 2013, 22, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Günther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Losa, D.; Chanson, M.; Crespin, S. Connexins as therapeutic targets in lung disease. Expert Opin. Ther. Targets 2011, 15, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Koval, M.; Billaud, M.; Straub, A.C.; Johnstone, S.R.; Zarbock, A.; Duling, B.R.; Isakson, B.E. Spontaneous lung dysfunction and fibrosis in mice lacking connexin 40 and endothelial cell connexin 43. Am. J. Pathol. 2011, 178, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Trovato-Salinaro, A.; Trovato-Salinaro, E.; Failla, M.; Mastruzzo, C.; Tomaselli, V.; Gili, E.; Crimi, N.; Condorelli, D.F.; Vancheri, C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 122. [Google Scholar] [CrossRef]
- Cesen-Cummings, K.; Fernstrom, M.J.; Malkinson, A.M.; Ruch, R.J. Frequent reduction of gap junctional intercellular communication and connexin43 expression in human and mouse lung carcinoma cells. Carcinogenesis 1998, 19, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Tourkina, E.; Richard, M.; Gööz, P.; Bonner, M.; Pannu, J.; Harley, R.; Bernatchez, P.N.; Sessa, W.C.; Silver, R.M.; Hoffman, S. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L843–L861. [Google Scholar] [CrossRef]
- Shivshankar, P.; Brampton, C.; Miyasato, S.; Kasper, M.; Thannickal, V.J.; Le Saux, C.J. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 28–36. [Google Scholar] [CrossRef]
- Senetta, R.; Stella, G.; Pozzi, E.; Sturli, N.; Massi, D.; Cassoni, P. Caveolin-1 as a promoter of tumour spreading: When, how, where and why. J. Cell. Mol. Med. 2013, 17, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Fischbach, M.A.; Haber, D.A.; Settleman, J. “Oncogenic Shock”: Explaining oncogene addiction through differential signal attenuation. Clin. Cancer Res. 2006, 12, 4392s–4395s. [Google Scholar] [CrossRef]
- Stella, G.M.; Inghilleri, S.; Pignochino, Y.; Zorzetto, M.; Oggionni, T.; Morbini, P.; Luisetti, M. Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl. Oncol. 2014, 7, 650–655. [Google Scholar] [CrossRef]
- Herbst, R.S.; Sandler, A.B. Overview of the current status of human epidermal growth factor receptor inhibitors in lung cancer. Clin. Lung Cancer 2004, 6, S7–S19. [Google Scholar] [CrossRef]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. 2004, 59, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Stella, G.M.; Luisetti, M.; Pozzi, E.; Comoglio, P.M. Oncogenes in non-small-cell lung cancer: Emerging connections and novel therapeutic dynamics. Lancet Respir. Med. 2013, 1, 251–261. [Google Scholar] [CrossRef]
- Veale, D.; Kerr, N.; Gibson, G.J.; Kelly, P.J.; Harris, A.L. The relationship of quantitative epidermal growth factor receptor expression in non-small cell lung cancer to long term survival. Br. J. Cancer 1993, 68, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoshiba, K.; Yokohori, N.; Nagai, A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003, 63, 5054–5059. [Google Scholar]
- Vallath, S.; Hynds, R.E.; Succony, L.; Janes, S.M.; Giangreco, A. Targeting EGFR signalling in chronic lung disease: Therapeutic challenges and opportunities. Eur. Respir. J. 2014, 44, 513–522. [Google Scholar] [CrossRef]
- Hardie, W.D.; Davidson, C.; Ikegami, M.; Leikauf, G.D.; Le Cras, T.D.; Prestridge, A.; Whitsett, J.A.; Korfhagen, T.R. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-α-induced pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1217–L1225. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. BioMed Res. Int. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Fujimoto, S.; Fukuda, T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, M.; Bachem, M.; Anders, D.; Trischler, G.; Faehling, M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 2005, 183, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.P.; Colby, T.V.; Travis, W.D.; Borkowski, A.; Jones, R.T.; Lane, D.P.; Metcalf, R.A.; Samet, J.M.; Takeshima, Y.; Gu, J.R.; et al. p53 protein accumulates frequently in early bronchial neoplasia. Cancer Res. 1993, 53, 4817–4822. [Google Scholar]
- Sozzi, G.; Miozzo, M.; Donghi, R.; Pilotti, S.; Cariani, C.T.; Pastorino, U.; Della Porta, G.; Pierotti, M.A. Deletions of 17p and p53 mutations in preneoplastic lesions of the lung. Cancer Res. 1992, 52, 6079–6082. [Google Scholar] [PubMed]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef]
- Kawasaki, H.; Ogura, T.; Yokose, T.; Nagai, K.; Nishiwaki, Y.; Esumi, H. p53 Gene alteration in atypical epithelial lesions and carcinoma in patients with idiopathic pulmonary fibrosis. Hum. Pathol. 2001, 32, 1043–1049. [Google Scholar] [CrossRef]
- Mascaux, C.; Martin, B.; Verdebout, J.; Meert, A.; Ninane, V.; Sculier, J. Fragile histidine triad protein expression in nonsmall cell lung cancer and correlation with Ki-67 and with p53. Eur. Respir. J. 2003, 21, 753–758. [Google Scholar] [CrossRef]
- Uematsu, K.; Yoshimura, A.; Gemma, A.; Mochimaru, H.; Hosoya, Y.; Kunugi, S.; Matsuda, K.; Seike, M.; Kurimoto, F.; Takenaka, K.; et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001, 61, 8527–8533. [Google Scholar]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Du Bois, R.M. A progression-free end-point for idiopathic pulmonary fibrosis trials: Lessons from cancer. Eur. Respir. J. 2012, 41, 262–269. [Google Scholar] [CrossRef]
- Vancheri, C. Idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 153–157. [Google Scholar] [CrossRef]
- Stella, G.M.; Balestro, E.; Lacedonia, D.; Baraldo, S. Telomeropathies: An emerging spectrum of disorders with important implications for patients with interstitial lung disease. Minerva Med. 2016, 107 (Suppl. 1), 9–14. [Google Scholar] [PubMed]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- De Leon, A.D.; Cronkhite, J.T.; Katzenstein, A.-L.A.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef]
- Parry, E.M.; Alder, J.K.; Qi, X.; Chen, J.J.-L.; Armanios, M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood 2011, 117, 5607–5611. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Samara, K.D.; Lasithiotaki, I.; Margaritopoulos, G.A.; Soufla, G.; Lambiri, I.; Giannarakis, I.; Drositis, I.; Spandidos, D.A.; Siafakas, N.M. Differential telomerase expression in idiopathic pulmonary fibrosis and non-small cell lung cancer. Oncol. Rep. 2013, 30, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Margaritopoulos, G.A.; Proklou, A.; Karagiannis, K.; Lasithiotaki, I.; Soufla, G.; Kastrinaki, M.C.; Spandidos, D.A.; Papadaki, H.A.; Siafakas, N.M. Investigation of telomerase/telomeres system in bone marrow mesenchymal stem cells derived from IPF and RA-UIP. J. Inflamm. 2012, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Lung, H.L.; Bangarusamy, D.K.; Xie, D.; Cheung, A.K.L.; Cheng, Y.; Kumaran, M.K.; Miller, L.; Liu, E.T.-B.; Guan, X.-Y.; Sham, J.S.; et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene 2005, 24, 6525–6532. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Kumbla, P.; Hagood, J.S. Enhanced myofibroblastic differentiation and survival in Thy-1(−) lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2007, 36, 226–235. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Pardo, A.; Selman, M.; Nuovo, G.J.; Tollefsbol, T.O.; Siegal, G.P.; Hagood, J.S. Thy-1 promoter hypermethylation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global Methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef]
- Gemma, A.; Takenaka, K.; Yoshimura, A.; Hosoya, Y.; Nara, M.; Hosomi, Y.; Okano, T.; Kunugi, S.; Koizumi, K.; Fukuda, Y.; et al. Reduced transcription of the Smad4 gene during pulmonary carcinogenesis in idiopathic pulmonary fibrosis. Mol. Med. Rep. 2008, 2, 73–80. [Google Scholar] [CrossRef]
- Langevin, S.M.; Kratzke, R.A.; Kelsey, K.T. Epigenetics of lung cancer. Transl. Res. 2015, 165, 74–90. [Google Scholar] [CrossRef]
- Huang, S.K.; Scruggs, A.M.; McEachin, R.C.; White, E.S.; Peters-Golden, M. Lung Fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in dna methylation compared to fibroblasts from nonfibrotic lung. PLoS ONE 2014, 9, e107055. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Yan, Q.; Chen, J.; Li, W.; Bao, C.; Fu, Q. Targeting miR-155 to treat experimental scleroderma. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Vancheri, C. Idiopathic pulmonary fibrosis and cancer: Do they really look similar? BMC Med. 2015, 13, 1–5. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; De Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in Pulmonary Fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E.H. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Shen, H.; Qiu, C.; Ni, Y.; Wang, L.; Dong, W.; Liao, Y.; Du, J. High expression of miR-21 and miR-155 predicts recurrence and unfavourable survival in non-small cell lung cancer. Eur. J. Cancer 2013, 49, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Eissa, M.G.; Artlett, C.M. The MicroRNA miR-155 Is essential in fibrosis. Non Coding RNA 2019, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Pparγ-modulated miRNA hubs that target the fibrotic tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Vukmirovic, M.; Kaminski, N. Impact of transcriptomics on our understanding of pulmonary fibrosis. Front. Med. 2018, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Arce, L.; Wang, M.; Putta, S.; Lanting, L.; Natarajan, R. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011, 80, 358–368. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.M.; Gabbiani, G. Recent developments in myofibroblast biology. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Landi, C.; Carleo, A.; Vantaggiato, L.; Bergantini, L.; d’Alessandro, M.; Cameli, P.; Sebastiani, G.; Dotta, F.; Bargagli, E. Common molecular pathways targeted by nintedanib in cancer and IPF: A bioinformatic study. Pulm. Pharmacol. Ther. 2020, 64, 101941. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Fois, S.S.; Fois, A.G.; Cossu, A.; Palmieri, G.; Pintus, G. Repurposing anticancer drugs for the treatment of idiopathic pulmonary fibrosis and antifibrotic drugs for the treatment of cancer: State of the art. Curr. Med. Chem. 2020, 27, 1–11. [Google Scholar] [CrossRef]
- Varone, F.; Sgalla, G.; Iovene, B.; Bruni, T.; Richeldi, L. Nintedanib for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 2018, 19, 167–175. [Google Scholar] [CrossRef]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Singh, S.R.; Hall, I.P. Airway myofibroblasts and their relationship with airway myocytes and fibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 127–132. [Google Scholar] [CrossRef]
- Dancer, R.C.A.; Wood, A.M.; Thickett, D.R. Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 1461–1467. [Google Scholar] [CrossRef]
- Niki, T.; Kohno, T.; Iba, S.; Moriya, Y.; Takahashi, Y.; Saito, M.; Maeshima, A.; Yamada, T.; Matsuno, Y.; Fukayama, M.; et al. frequent co-localization of Cox-2 and Laminin-5 γ2 Chain at the invasive front of early-stage lung adenocarcinomas. Am. J. Pathol. 2002, 160, 1129–1141. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Skacel, M.; Adams, J.C. Roles of fascin in human carcinoma motility and signaling: Prospects for a novel biomarker? Int. J. Biochem. Cell Biol. 2005, 37, 1787–1804. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Bogatkevic, G.S.; Atnelishvili, I.; Akter, T.; Feghali-Bostwick, C.; Hoffman, S.; Fresco, V.M.; Fucsh, J.V.; Visconti, R.P.; Markwald, R.R.; et al. Overexpression of c-MET and CD44v6 receptors contributes to autocrine TGF-β1 signaling in interstitial lung disease. J. Biol. Chem. 2014, 289, 7856–7857. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-Catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Shi, J.; Li, F.; Luo, M.; Wei, J.; Liu, X. Distinct Roles of Wnt/β-catenin signaling in the pathogenesis of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Mediat. Inflamm. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.H.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E.M. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, A.L.; Milla, C.M.; Lira, J.C.; Ramírez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of sonic hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog signaling in lung cancer: From Oncogenesis to cancer treatment resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef]
- Morimoto, M.; Liu, Z.; Cheng, H.-T.; Winters, N.; Bader, D.; Kopan, R. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J. Cell Sci. 2010, 123, 213–224. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Zou, B.; Zhou, X.; Lai, S.; Liu, J. Notch signaling and non-small cell lung cancer (Review). Oncol. Lett. 2018, 15, 3415–3421. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. 2013, 93, 566–576. [Google Scholar] [CrossRef]
- Wei, X.; Han, J.; Chen, Z.-Z.; Qi, B.-W.; Wang, G.-C.; Ma, Y.-H.; Zheng, H.; Luo, Y.-F.; Wei, Y.-Q.; Chen, L.-J. A phosphoinositide 3-kinase-γ inhibitor, AS605240 prevents bleomycin-induced pulmonary fibrosis in rats. Biochem. Biophys. Res. Commun. 2010, 397, 311–317. [Google Scholar] [CrossRef]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Platé, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Manuela, P.; Genovese, F.; et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; Fahy, W.A.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef]
- D’Alessandro, M.; Bergantini, L.; Torricelli, E.; Cameli, P.; Lavorini, F.; Pieroni, M.; Refini, R.; Sestini, P.; Bargagli, E. Systematic review and metanalysis of oncomarkers in IPF patients and serial changes of oncomarkers in a prospective italian real-life case series. Cancers 2021, 13, 539. [Google Scholar] [CrossRef]
- Stock, C.J.; Hoyles, R.K.; Daccord, C.; Kokosi, M.; Visca, D.; De Lauretis, A.; Alfieri, V.; Kouranos, V.; Margaritopoulos, G.; George, P.M.; et al. Serum markers of pulmonary epithelial damage in systemic sclerosis-associated interstitial lung disease and disease progression. Respirology 2020. [Google Scholar] [CrossRef]
- Balestro, E.; Castelli, G.; Bernardinello, N.; Cocconcelli, E.; Biondini, D.; Fracasso, F.; Rea, F.; Saetta, M.; Baraldo, S.; Spagnolo, P. CA 19-9 serum levels in patients with end-stage idiopathic pulmonary fibrosis (IPF) and other interstitial lung diseases (ILDs): Correlation with functional decline. Chronic Respir. Dis. 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; Bargagli, E.; Cameli, P.; Cekorja, B.; Lanzarone, N.; Pianigiani, L.; Vietri, L.; Bennett, D.; Sestini, P.; Rottoli, P. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir. Investig. 2019, 57, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Moll, S.A.; Wiertz, I.A.; Vorselaars, A.D.; Zanen, P.; Ruven, H.J.; Van Moorsel, C.H.; Grutters, J.C. Serum biomarker CA 15-3 as predictor of response to antifibrotic treatment and survival in idiopathic pulmonary fibrosis. Biomark. Med. 2020, 14, 997–1007. [Google Scholar] [CrossRef]
- Ueda, T.; Aokage, K.; Nishikawa, H.; Neri, S.; Nakamura, H.; Sugano, M.; Tane, K.; Miyoshi, T.; Kojima, M.; Fujii, S.; et al. Immunosuppressive tumor microenvironment of usual interstitial pneumonia-associated squamous cell carcinoma of the lung. J. Cancer Res. Clin. Oncol. 2018, 144, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Aokage, K.; Mimaki, S.; Tane, K.; Miyoshi, T.; Sugano, M.; Kojima, M.; Fujii, S.; Kuwata, T.; Ochiai, A.; et al. Characterization of the tumor immune-microenvironment of lung adenocarcinoma associated with usual interstitial pneumonia. Lung Cancer 2018, 126, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Aokage, K.; Neri, S.; Nakamura, H.; Nomura, S.; Tane, K.; Miyoshi, T.; Sugano, M.; Kojima, M.; Fujii, S.; et al. Link between tumor-promoting fibrous microenvironment and an immunosuppressive microenvironment in stage I lung adenocarcinoma. Lung Cancer 2018, 126, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-L.; Ni, C.-F.; Liang, H.-S.; Xu, Y.-H.; Wang, W.-S.; Shen, J.; Li, M.-M.; Zhu, X.-L. Upregulation of PD-L1 expression promotes epithelial-to-mesenchymal transition in sorafenib-resistant hepatocellular carcinoma cells. Gastroenterol. Rep. 2020, 8, 390–398. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Crispen, P.L.; Kusmartsev, S. Mechanisms of immune evasion in bladder cancer. Cancer Immunol. Immunother. 2020, 69, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef]
- Lo, U.-G.; Pong, R.-C.; Yang, D.; Gandee, L.; Hernandez, E.; Dang, A.; Lin, C.-J.; Santoyo, J.; Ma, S.; Sonavane, R.; et al. IFN-r-induced IFIT5 promotes epithelial-to-mesenchymal transition in prostate cancer via microRNA processing. Cancer Res. 2018, 79, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ma, Q. In Vivo Activation and Pro-Fibrotic Function of NF-κB in Fibroblastic cells during pulmonary inflammation and fibrosis induced by carbon nanotubes. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Lihui, H.; Ji, H.; Zhao, B.; Xu, C.; Xia, W.; Han, L.; Yu, D.; Ju, Y.; Jin, C. Therapeutic effect of ulinastatin on pulmonary fibrosis via downregulation of TGF-β1, TNF-α and NF-κB. Mol. Med. Rep. 2017, 17, 1717–1723. [Google Scholar] [CrossRef]
- Rahmani, F.; Asgharzadeh, F.; Avan, A.; Barneh, F.; Parizadeh, M.R.; Ferns, G.A.; Ryzhikov, M.; Ahmadian, M.R.; Giovannetti, E.; Jafari, M.; et al. Rigosertib potently protects against colitis-associated intestinal fibrosis and inflammation by regulating PI3K/AKT and NF-κB signaling pathways. Life Sci. 2020, 249, 117470. [Google Scholar] [CrossRef]
- Segel, M.J.; Izbicki, G.; Cohen, P.Y.; Or, R.; Christensen, T.G.; Wallach-Dayan, S.B.; Breuer, R. Role of interferon-γ in the evolution of murine bleomycin lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L1255–L1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.S.; Greenlee, B.M.; Wills-Karp, M.; Moller, D.R. Attenuation of lung inflammation and fibrosis in interferon- γ –Deficient Mice after intratracheal bleomycin. Am. J. Respir. Cell Mol. Biol. 2001, 24, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.N.; Chen, X.; Foda, H.D.; Smaldone, G.C.; Hasaneen, N.A. Interferon-γ enhances the antifibrotic effects of pirfenidone by attenuating IPF lung fibroblast activation and differentiation. Respir. Res. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- King, T.E.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune microenvironment profiling of gastrointestinal stromal tumors (GIST) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology 2019, 8, e1617588. [Google Scholar] [CrossRef]
- Dezutter-Dambuyant, C.; Durand, I.; Alberti, L.; Bendriss-Vermare, N.; Valladeau-Guilemond, J.; Duc, A.; Magron, A.; Morel, A.-P.; Sisirak, V.; Rodriguez, C.; et al. A novel regulation of PD-1 ligands on mesenchymal stromal cells through MMP-mediated proteolytic cleavage. Oncoimmunology 2016, 5, e1091146. [Google Scholar] [CrossRef]
- Beswick, E.J.; Johnson, J.R.; Saada, J.I.; Humen, M.; House, J.; Dann, S.; Qiu, S.; Brasier, A.R.; Powell, D.W.; Reyes, V.E.; et al. TLR4 Activation Enhances the PD-L1–Mediated Tolerogenic Capacity of Colonic CD90+ Stromal Cells. J. Immunol. 2014, 193, 2218–2229. [Google Scholar] [CrossRef]
- Zhao, J.; Xiao, Z.; Li, T.; Chen, H.; Yuan, Y.; Wang, Y.A.; Hsiao, C.-H.; Chow, D.S.-L.; Overwijk, W.W.; Li, C. Stromal modulation reverses primary resistance to immune checkpoint blockade in pancreatic cancer. ACS Nano 2018, 12, 9881–9893. [Google Scholar] [CrossRef]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Vrishika, K.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Jovanovic, D.; Milenkovic, M.R.; Stevuljevic, J.K.; Markovic, J.; Ceriman, V.; Kontic, M.; Trifunovic, V.S. Membrane PD-L1 expression and soluble PD-L1 plasma levels in idiopathic pulmonary fibrosis—A pilot study. J. Thorac. Dis. 2018, 10, 6660–6669. [Google Scholar] [CrossRef]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.-T.; Liu, Y.; Chan, G.C.-F.; Lau, Y.-L.; et al. PD-1/PD-L1 Pathway Mediates the alleviation of pulmonary fibrosis by human mesenchymal stem cells in humanized mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef]
- Wang, B.; Bai, W.; Ma, H.; Li, F. Regulatory Effect of PD1/PD-Ligand 1 (PD-L1) on Treg cells in patients with idiopathic pulmonary fibrosis. Med. Sci. Monit. 2020, 26, e927577-1. [Google Scholar] [CrossRef]
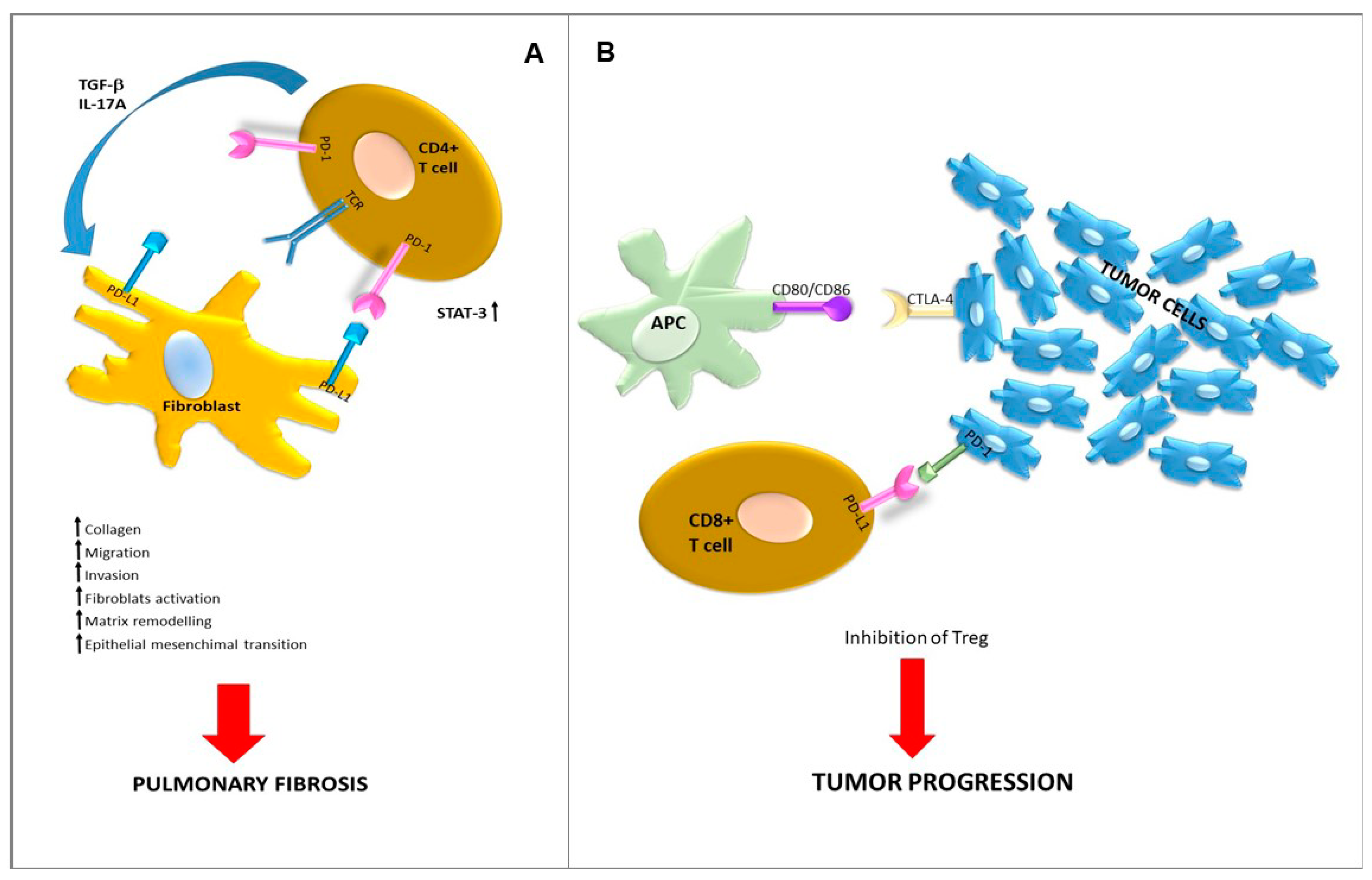
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef]
- Duitman, J.; Ende, T.V.D.; Spek, C.A. Immune checkpoints as promising targets for the treatment of idiopathic pulmonary fibrosis? J. Clin. Med. 2019, 8, 1547. [Google Scholar] [CrossRef]
- Habiel, D.M.; Espindola, M.S.; Kitson, C.; Azzara, A.V.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. Characterization of CD28 null T cells in idiopathic pulmonary fibrosis. Mucosal Immunol. 2018, 12, 212–222. [Google Scholar] [CrossRef]
- Justet, A.; Laurent-Bellue, A.; Thabut, G.; Dieudonné, A.; Debray, M.-P.; Borie, R.; Aubier, M.; Lebtahi, R.; Crestani, B. [18F]FDG PET/CT predicts progression-free survival in patients with idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 1–10. [Google Scholar] [CrossRef]
- Désogère, P.; Tapias, L.F.; Rietz, T.A.; Rotile, N.; Blasi, F.; Day, H.; Elliott, J.; Fuchs, B.C.; Lanuti, M.; Caravan, P. Optimization of a Collagen-Targeted PET Probe for molecular imaging of pulmonary fibrosis. J. Nucl. Med. 2017, 58, 1991–1996. [Google Scholar] [CrossRef]
- Kimura, R.H.; Wang, L.; Shen, B.; Huo, L.; Tummers, W.; Filipp, F.V.; Guo, H.H.; Haywood, T.; Abou-Elkacem, L.; Baratto, L.; et al. Evaluation of integrin αvβ6 cystine knot PET tracers to detect cancer and idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Biederer, J. Using lung mri and elastic registration to assess pulmonary fibrosis. Radiology 2019, 291, 493–494. [Google Scholar] [CrossRef]
- Lonzetti, L.; Zanon, M.; Pacini, G.S.; Altmayer, S.; de Oliveira, D.M.; Rubin, A.S.; Gazzoni, F.F.; Barros, M.C.; Hochhegger, B. Magnetic resonance imaging of interstitial lung diseases: A state-of-the-art review. Respir. Med. 2019, 155, 79–85. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lettieri, S.; Oggionni, T.; Lancia, A.; Bortolotto, C.; Stella, G.M. Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? Int. J. Mol. Sci. 2021, 22, 2882. https://doi.org/10.3390/ijms22062882
Lettieri S, Oggionni T, Lancia A, Bortolotto C, Stella GM. Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? International Journal of Molecular Sciences. 2021; 22(6):2882. https://doi.org/10.3390/ijms22062882
Chicago/Turabian StyleLettieri, Sara, Tiberio Oggionni, Andrea Lancia, Chandra Bortolotto, and Giulia Maria Stella. 2021. "Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape?" International Journal of Molecular Sciences 22, no. 6: 2882. https://doi.org/10.3390/ijms22062882
APA StyleLettieri, S., Oggionni, T., Lancia, A., Bortolotto, C., & Stella, G. M. (2021). Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? International Journal of Molecular Sciences, 22(6), 2882. https://doi.org/10.3390/ijms22062882