
Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods

Abstract

1. Introduction
2. Results
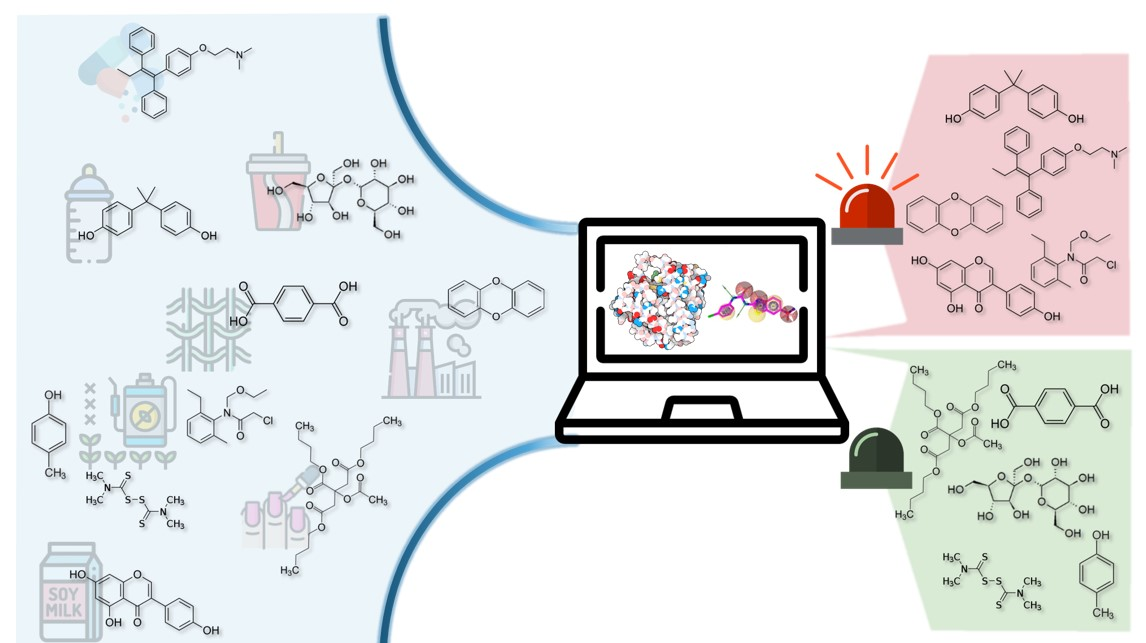
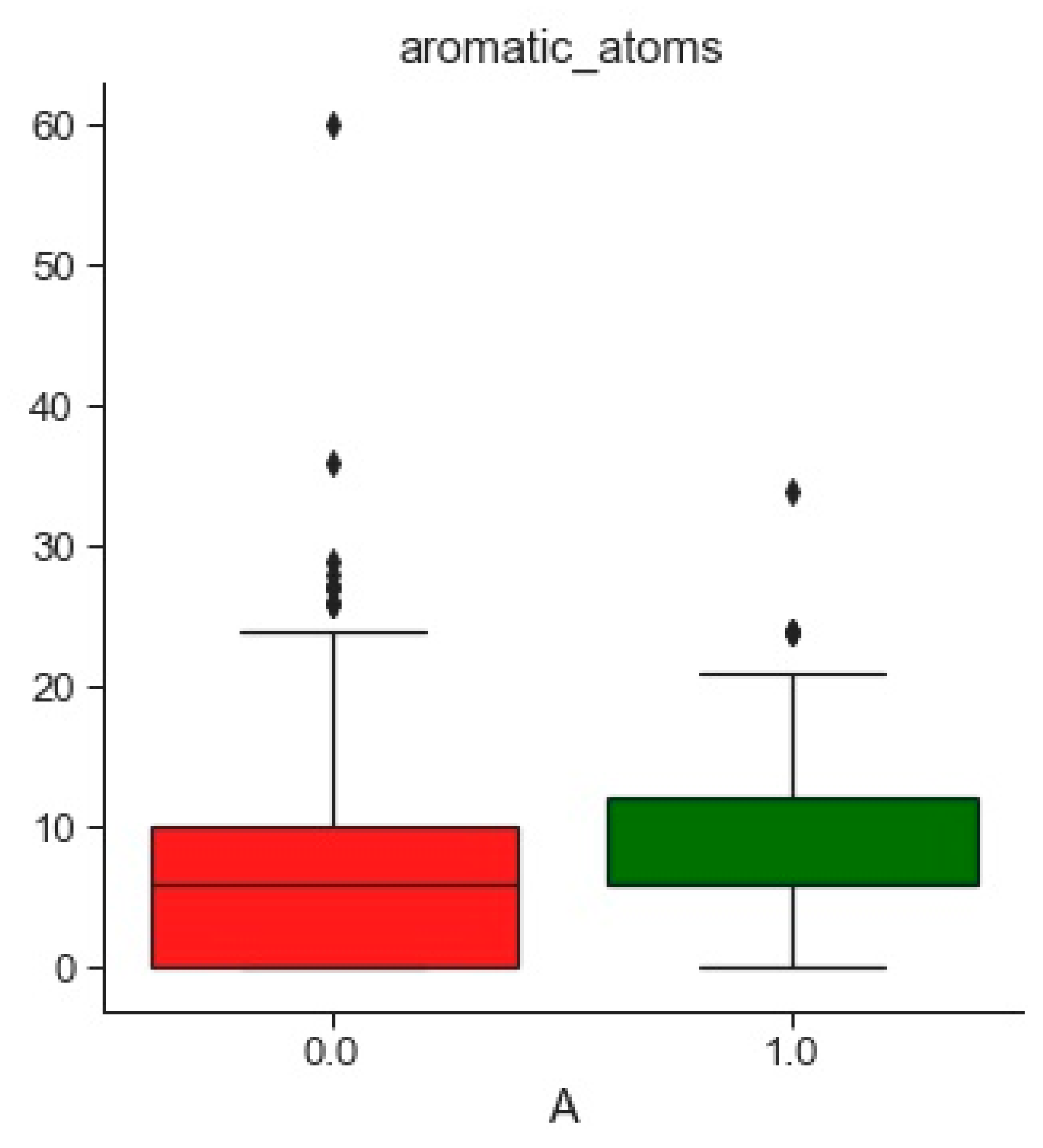
2.1. Compounds and Database Preparation
2.1.1. Database Preparation
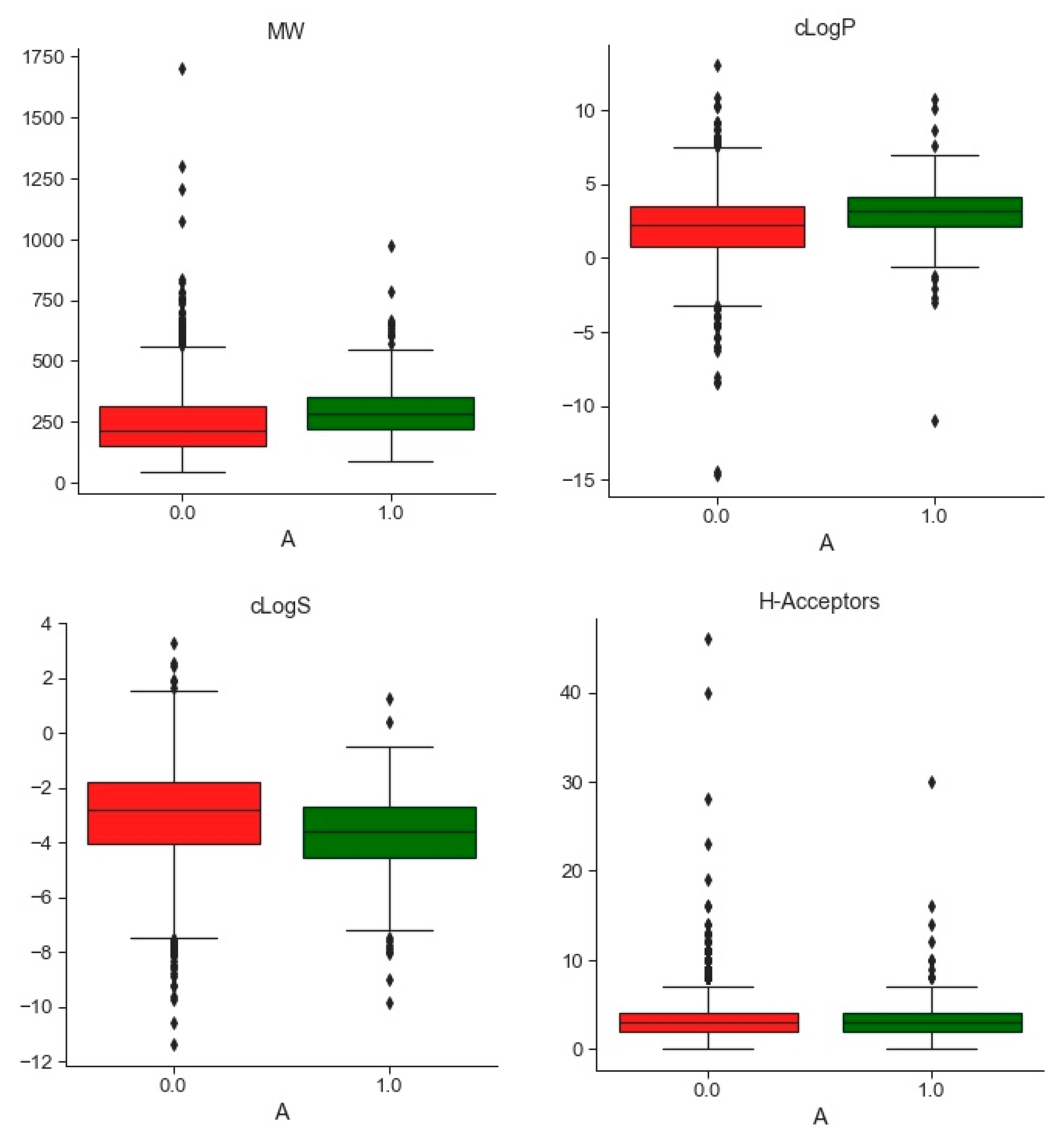
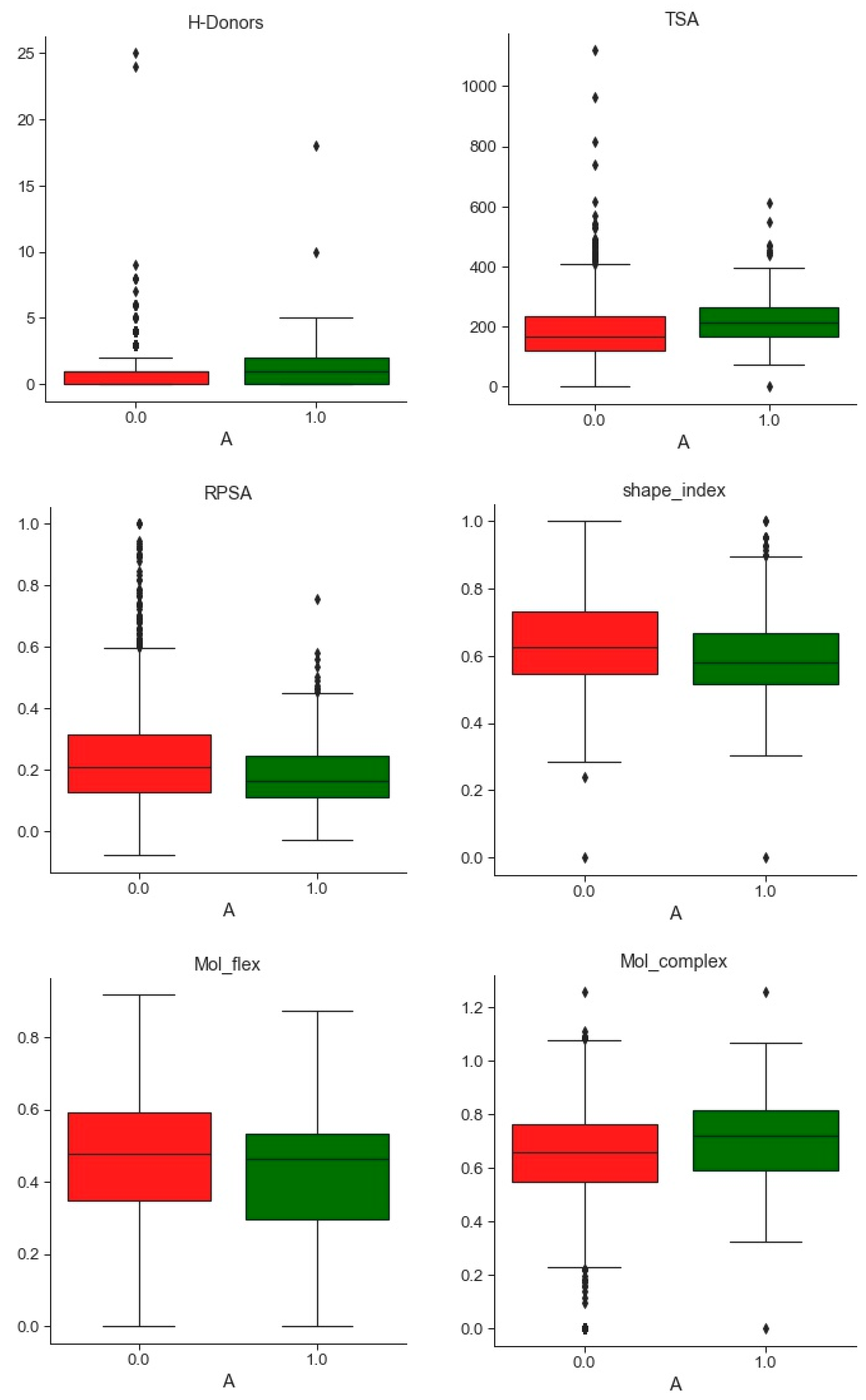
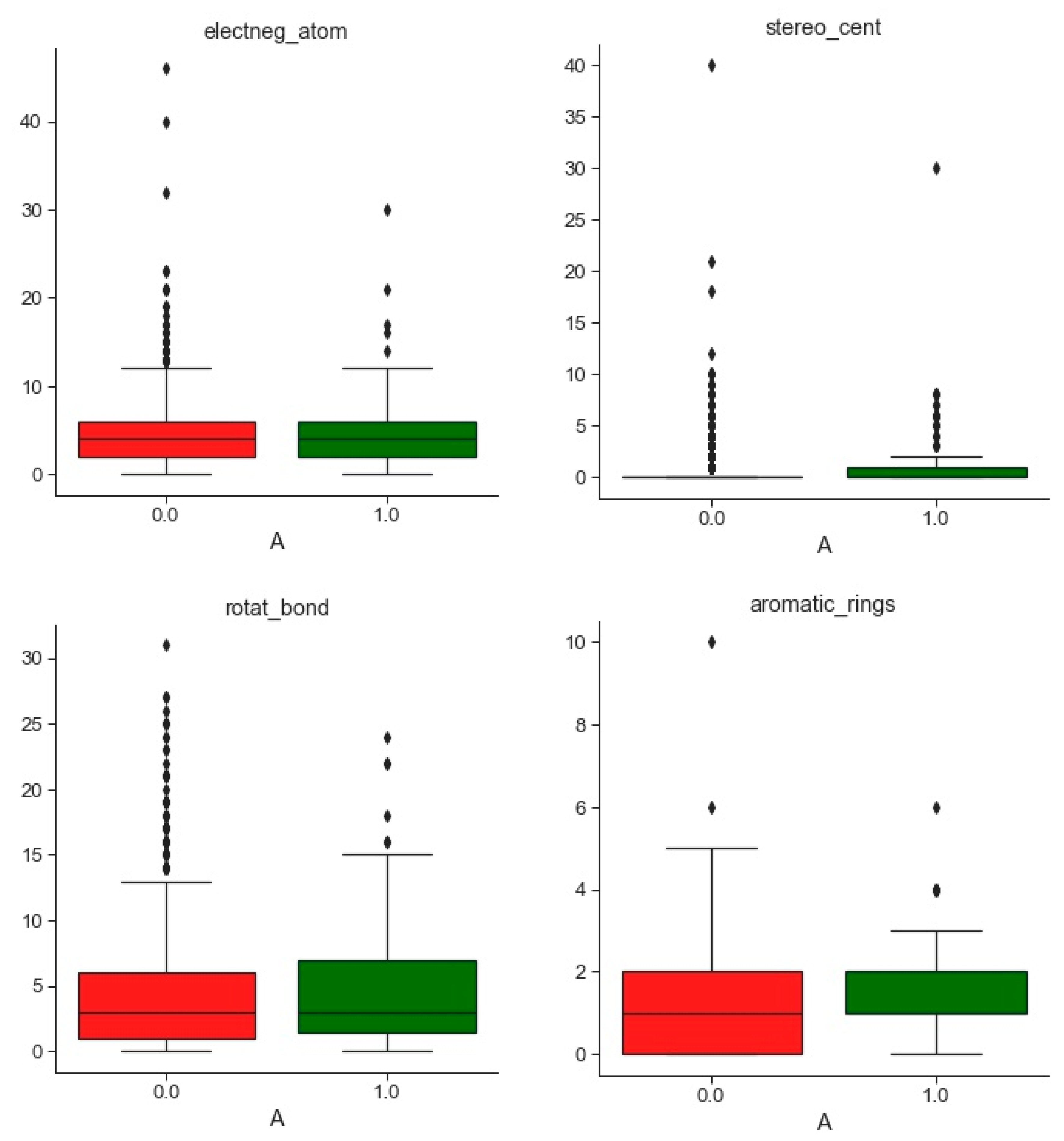
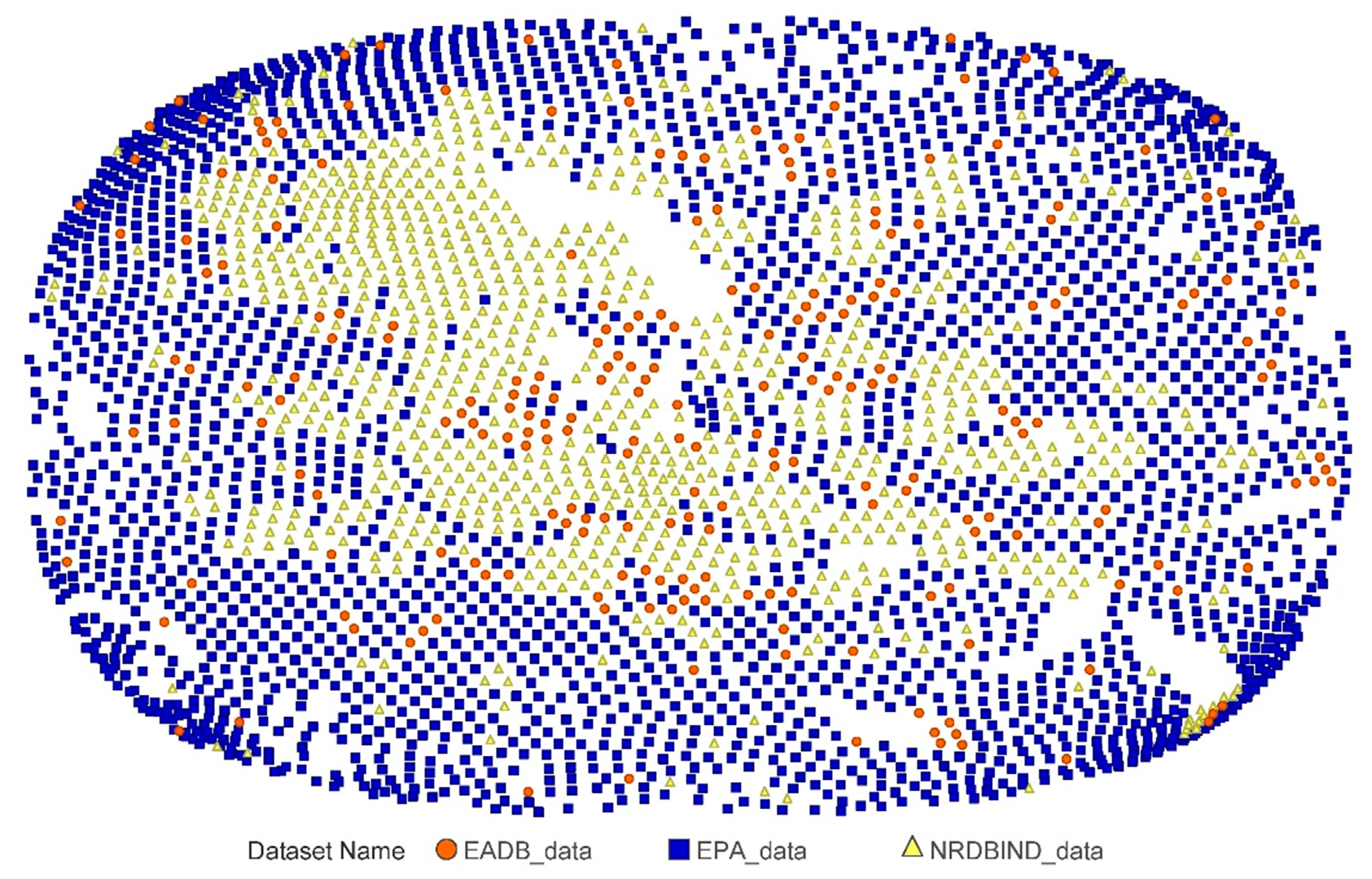
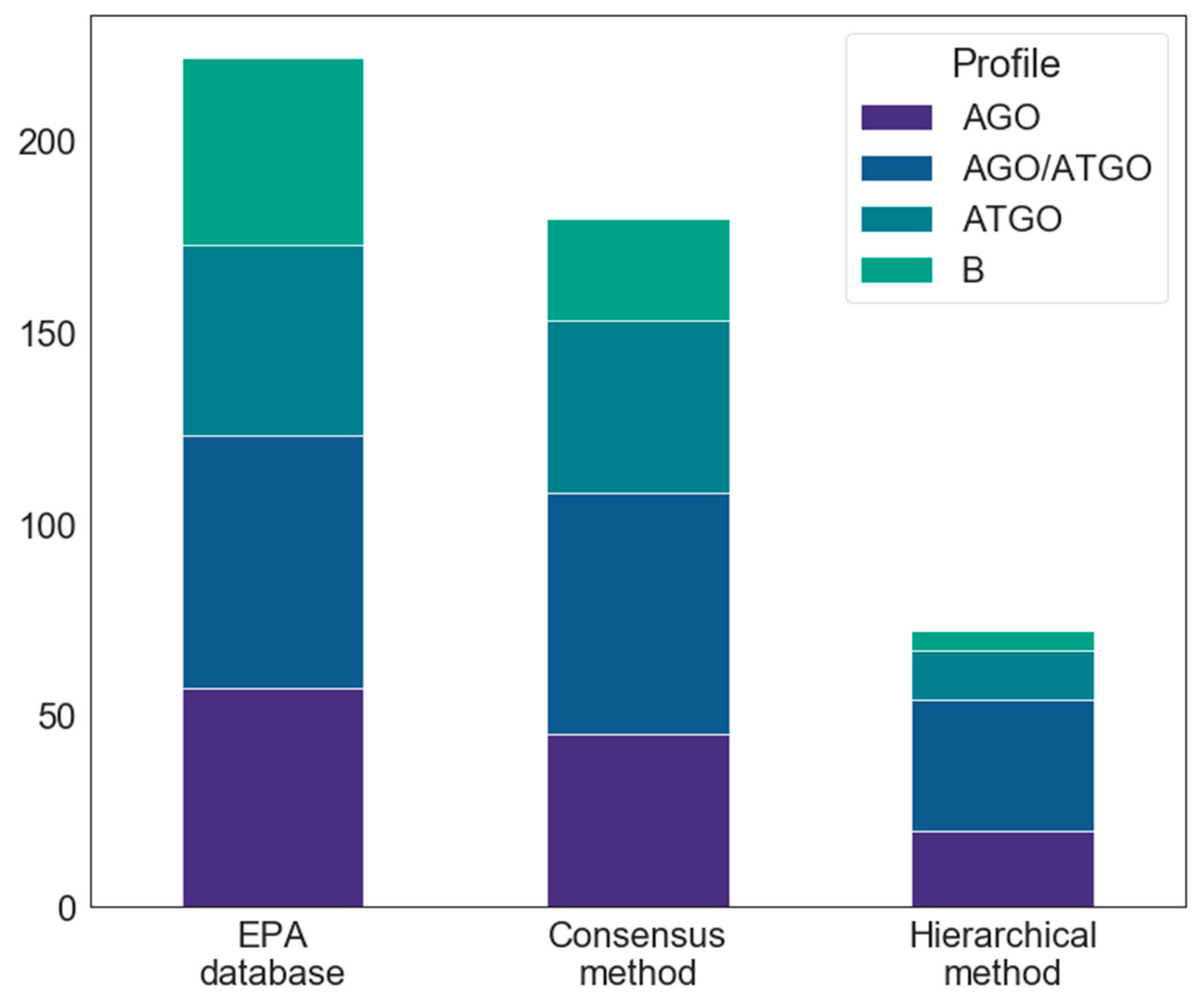
2.1.2. Databases Comparison
2.2. Docking
2.2.1. Docking Outcome
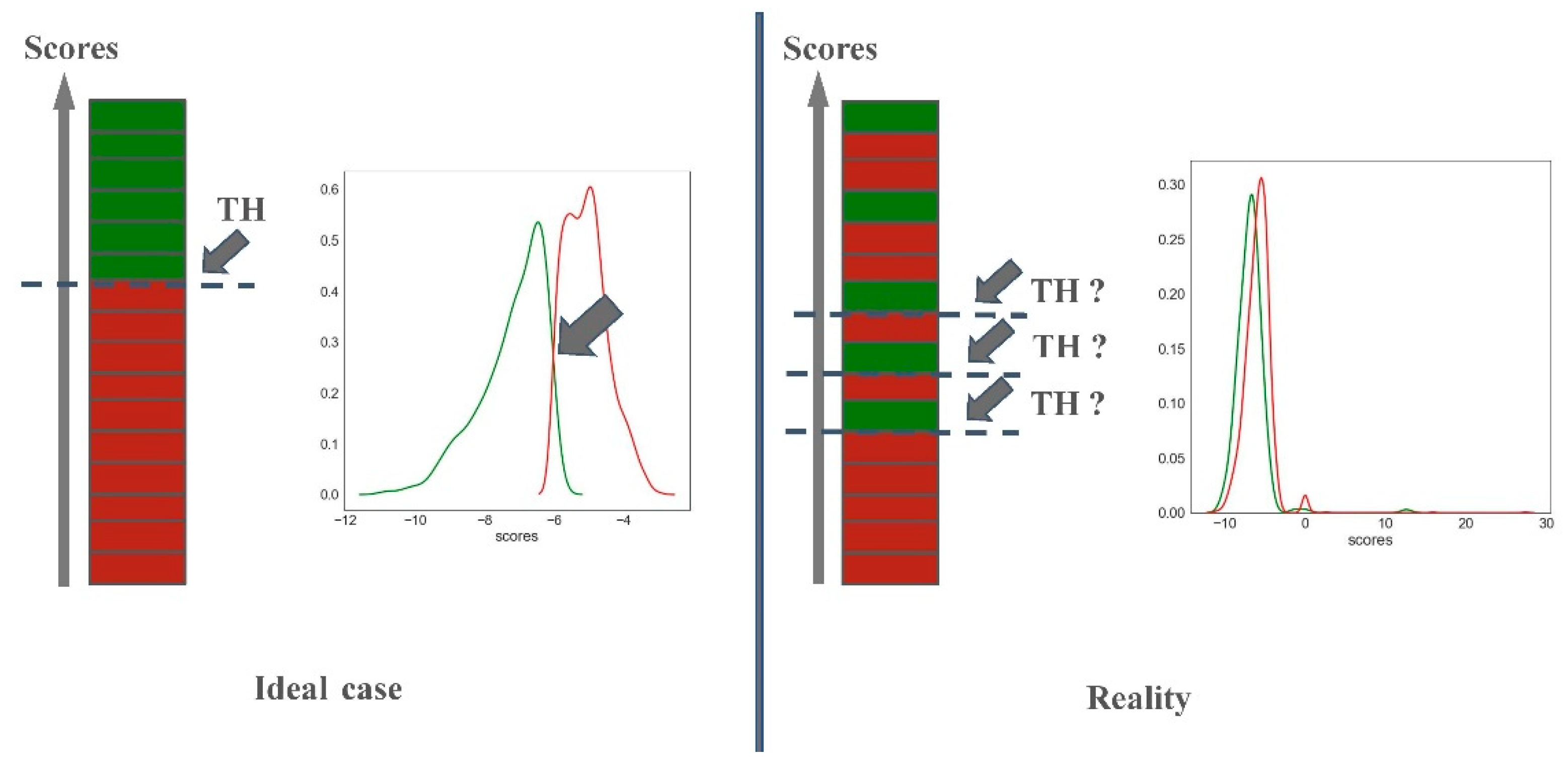
2.2.2. Predictiveness Curve
2.3. Pharmacophore Modeling
2.3.1. LB Pharmacophore Models
2.3.2. SB Pharmacophore Models
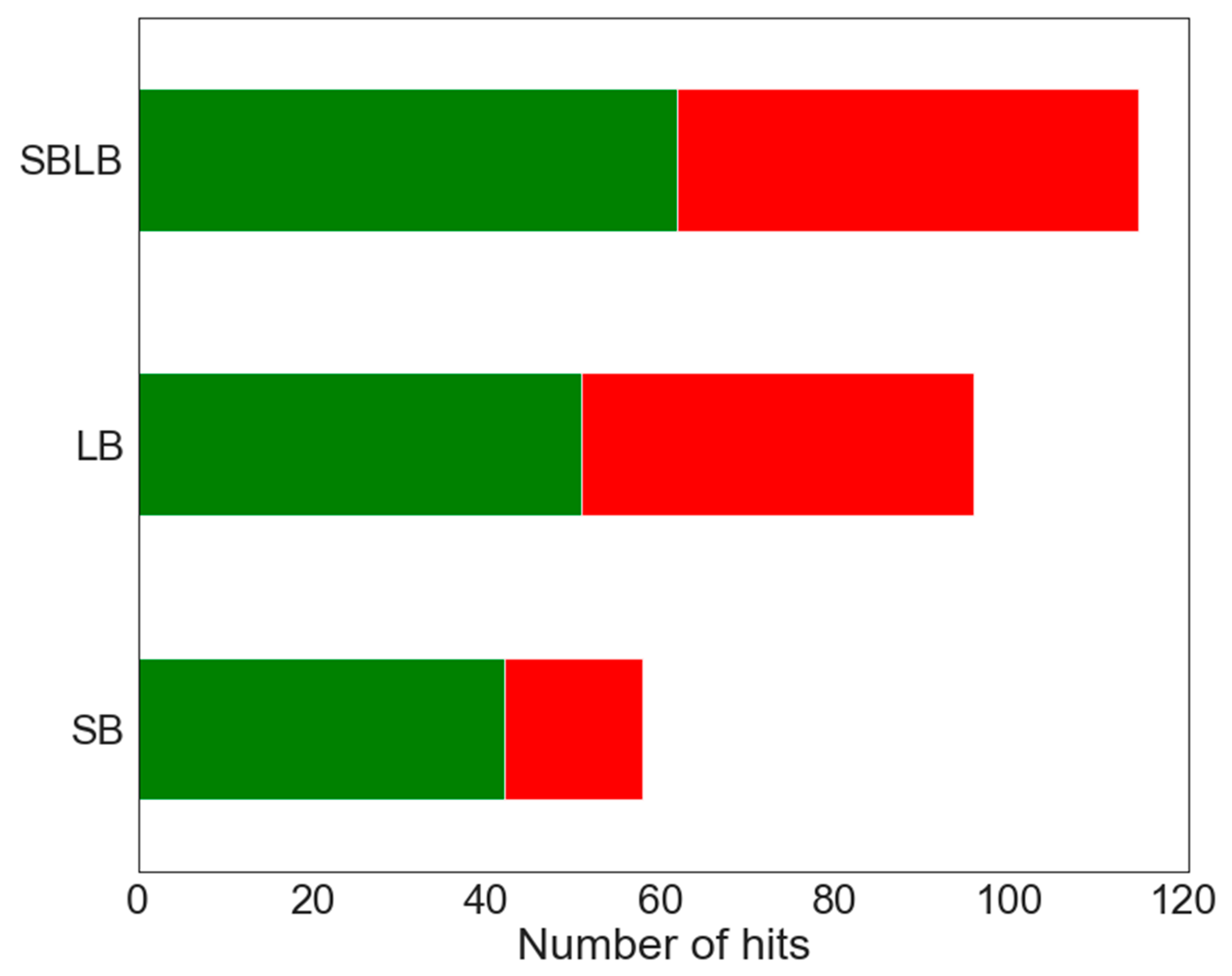
2.3.3. SBLB Pharmacophore Models
2.4. Combination of Docking and Pharmacophore Models
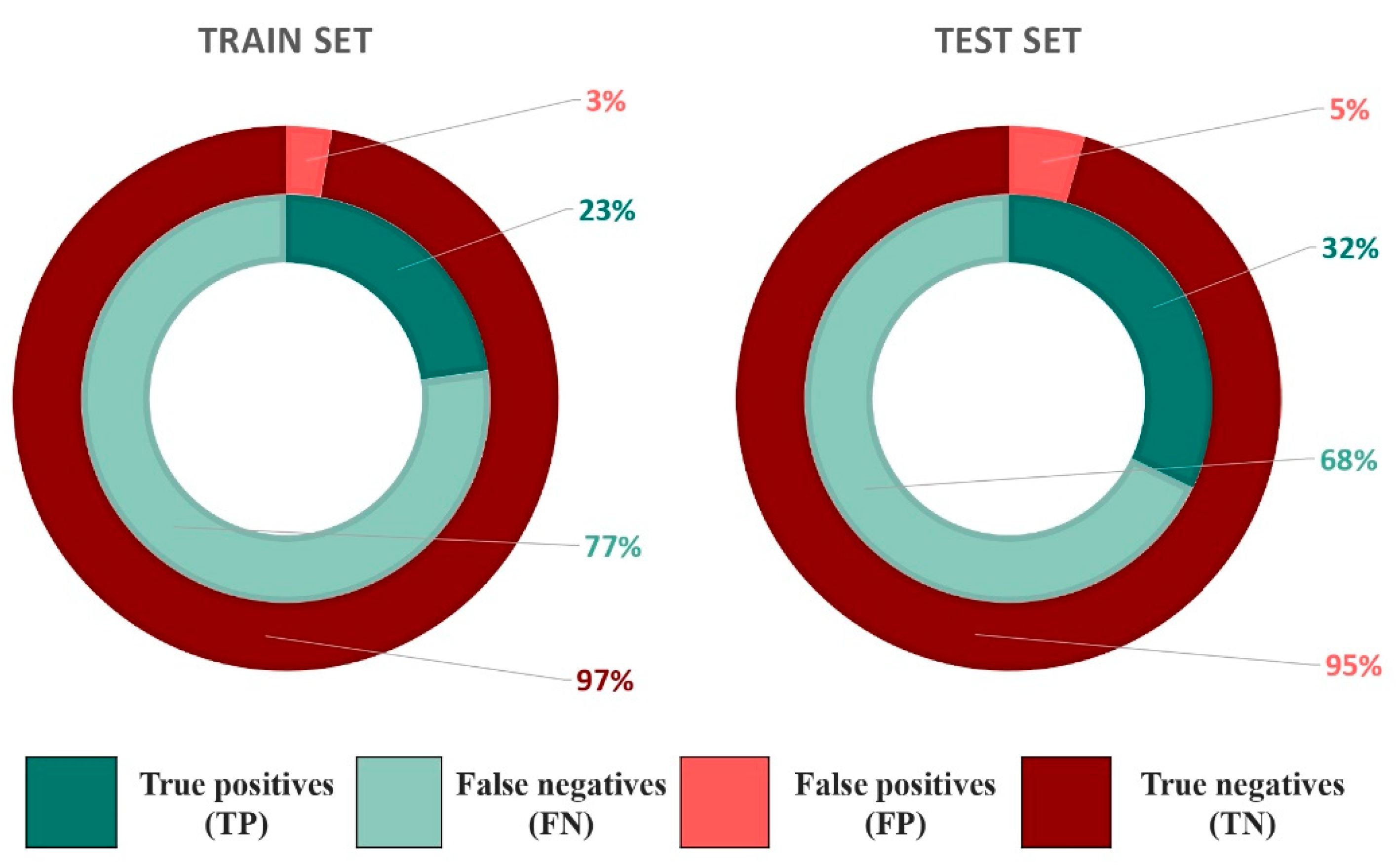
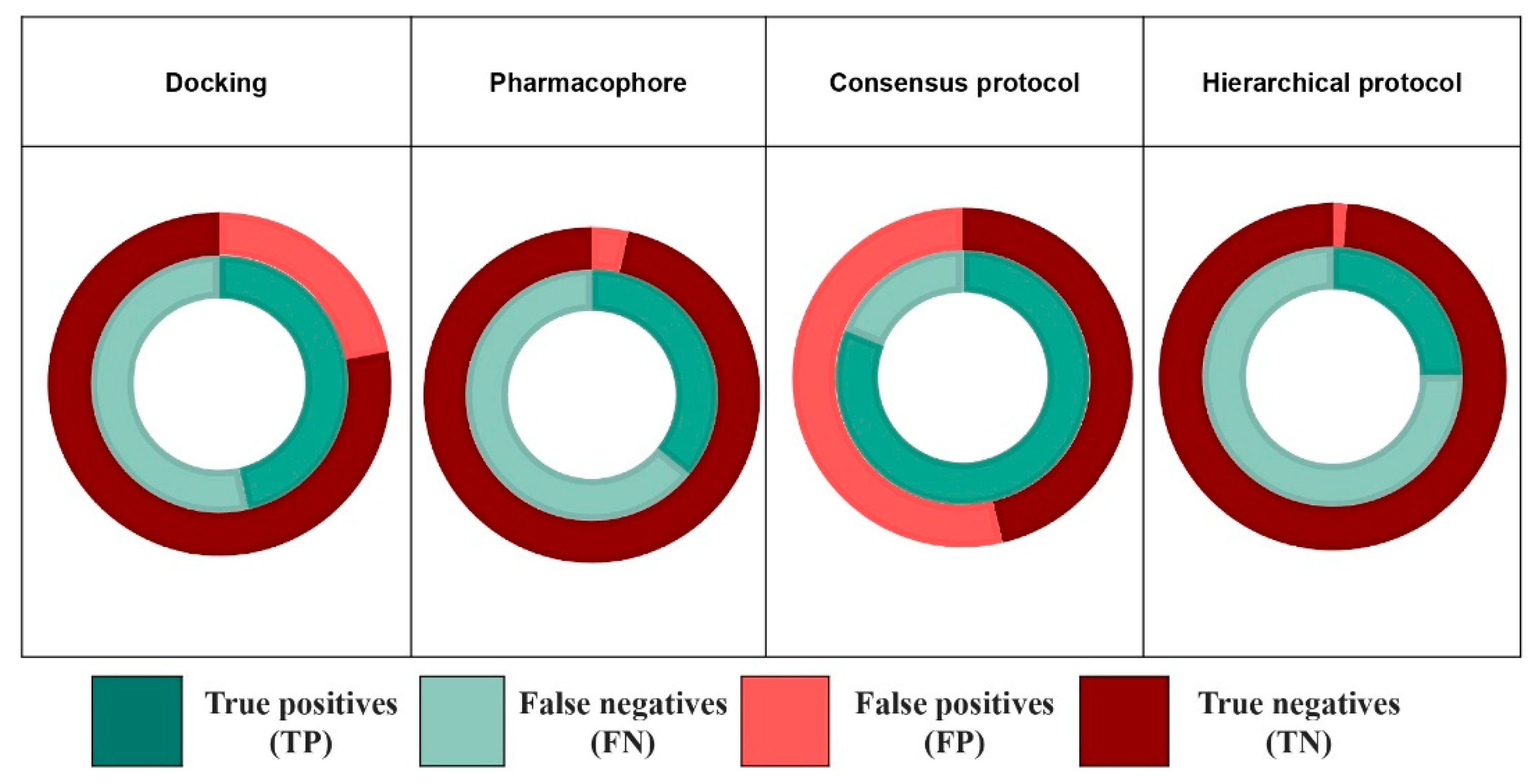
2.4.1. Consensus Protocol
2.4.2. Hierarchical Protocol
3. Discussion
3.1. Compounds and Database Preparation
3.2. Docking
3.3. Predictiveness Curve
3.4. Pharmacophores
3.5. Combination of Methods
4. Materials and Methods
4.1. Compounds, Databases Preparation, and Annotation
4.1.1. EPA Dataset
4.1.2. Validation Sets
- NR-DBIND
- EADB
4.1.3. Molecule Curation and Preparation
4.2. Structures Preparation
4.3. Docking
4.3.1. Protocol
4.3.2. Docking Performances Analyses
- Single structure docking and ensemble docking
- Predictiveness curves
4.4. Pharmacophore Modeling Protocol
4.4.1. Ligand Based Approach (LB) Models Protocol
4.4.2. Structure Based Approach (SB) Models Protocol
- Pharmacophore model optimization
4.4.3. Combination of SB and LB Pharmacophores Models
4.5. Pipelines Construction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptors |
| AUC | Area under the ROC curve |
| B | Binding compounds |
| CAS | Chemical Abstracts Service |
| DBD | DNA-binding domain |
| DNA | deoxyribonucleic acid |
| DSSTox | Distributed Structure-Searchable Toxicity |
| EADB | Estrogenic activity database |
| EDCs | Endocrine disrupting chemicals |
| EF | Enrichment factor |
| EPA | United states Environmental protection agency |
| ER | Estrogen receptors |
| FIX | Factor IX |
| GR | Glucocorticoid receptors |
| LB | Ligand based |
| LBD | Ligand binding domain |
| NB | Non-Binding compounds |
| NCTR | National center for toxicological research USA |
| NR | Nuclear receptor |
| NR-DBIND | Nuclear Receptors Database Including Negative Data |
| NTD | NH2-terminal domain |
| PC | Predictiveness curve |
| PDB | Protein data bank |
| PPAR | Peroxisome proliferator-activated receptors |
| PPV | Positive Predictive value |
| PLANTS | Protein-ligand ANTSystem |
| QSAR | Quantitative structure activity relationship |
| RMSD | Root-mean-square deviation |
| ROC | Receiver operating curve |
| SB | Structure based |
| SD | Standard deviation |
| Se | Sensitivity |
| SMILES | Simplified molecular-input line-entry system |
| Sp | Specificity |
| TH | scoring Threshold |
| TR | Thyroid hormones receptors |
References
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.-Å.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nat. Cell Biol. 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.Å. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Matthews, J.; Gustafsson, J.-A. Estrogen Signaling: A Subtle Balance between ER Alpha and ER Beta. Mol. Interv. 2003, 3, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, D.M.; Wang, Y.; Williams, S.P.; Sigler, P.B. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5998–6003. [Google Scholar] [CrossRef]
- Shao, W.; Brown, M. Advances in estrogen receptor biology: Prospects for improvements in targeted breast cancer therapy. Breast Cancer Res. 2003, 6, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, F.; Macchia, M.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor β ligands: Recent advances and biomedical applications. Med. Res. Rev. 2009, 31, 364–442. [Google Scholar] [CrossRef]
- Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction/Molecular Pharmaceutics. Available online: https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.8b00546 (accessed on 2 November 2020).
- Golden, R.J.; Noller, K.L.; Titus-Ernstoff, L.; Kaufman, R.H.; Mittendorf, R.; Stillman, R.; Reese, E.A. Environmental Endocrine Modulators and Human Health: An Assessment of the Biological Evidence. Crit. Rev. Toxicol. 1998, 28, 109–227. [Google Scholar] [CrossRef]
- Schug, T.T.; Johnson, A.F.; Birnbaum, L.S.; Colborn, T.; Guillette, L.J., Jr.; Crews, D.P.; Collins, T.; Soto, A.M.; vom Saal, F.S.; McLachlan, J.A.; et al. Minireview: Endocrine Disruptors: Past Lessons and Future Directions. Mol. Endocrinol. 2016, 30, 833–847. [Google Scholar] [CrossRef]
- Fillol, C.; Oleko, A.; Saoudi, A.; Zeghnoun, A.; Balicco, A.; Gane, J.; Rambaud, L.; Leblanc, A.; Gaudreau, É.; Marchand, P.; et al. Exposure of the French population to bisphenols, phthalates, parabens, glycol ethers, brominated flame retardants, and perfluorinated compounds in 2014–2016: Results from the Esteban study. Environ. Int. 2021, 147, 106340. [Google Scholar] [CrossRef] [PubMed]
- Audouze, K.; Sarigiannis, D.; Alonso-Magdalena, P.; Brochot, C.; Casas, M.; Vrijheid, M.; Babin, P.J.; Karakitsios, S.; Coumoul, X.; Barouki, R. Integrative Strategy of Testing Systems for Identification of Endocrine Disruptors Inducing Metabolic Disorders—An Introduction to the OBERON Project. Int. J. Mol. Sci. 2020, 21, 2988. [Google Scholar] [CrossRef]
- Johansson, H.K.L.; Svingen, T.; Fowler, P.A.; Vinggaard, A.M.; Boberg, J. Environmental influences on ovarian dysgenesis—Developmental windows sensitive to chemical exposures. Nat. Rev. Endocrinol. 2017, 13, 400–414. [Google Scholar] [CrossRef]
- Ghassabian, A.; Trasande, L. Disruption in Thyroid Signaling Pathway: A Mechanism for the Effect of Endocrine-Disrupting Chemicals on Child Neurodevelopment. Front. Endocrinol. 2018, 9, 204. [Google Scholar] [CrossRef]
- Cano-Sancho, G.; Salmon, A.G.; La Merrill, M.A. Association between Exposure to p,p′-DDT and Its Metabolite p,p′-DDE with Obesity: Integrated Systematic Review and Meta-Analysis. Environ. Health Perspect. 2017, 125, 096002. [Google Scholar] [CrossRef]
- Kumar, M.; Sarma, D.K.; Shubham, S.; Kumawat, M.; Verma, V.; Prakash, A.; Tiwari, R. Environmental Endocrine-Disrupting Chemical Exposure: Role in Non-Communicable Diseases. Front. Public Health 2020, 8, 553850. [Google Scholar] [CrossRef] [PubMed]
- Shanle, E.K.; Xu, W. Endocrine Disrupting Chemicals Targeting Estrogen Receptor Signaling: Identification and Mechanisms of Action. Chem. Res. Toxicol. 2010, 24, 6–19. [Google Scholar] [CrossRef]
- Combarnous, Y.; Nguyen, T.M.D. Comparative Overview of the Mechanisms of Action of Hormones and Endocrine Disruptor Compounds. Toxics 2019, 7, 5. [Google Scholar] [CrossRef]
- Balaguer, P.; Delfosse, V.; Bourguet, W. Mechanisms of endocrine disruption through nuclear receptors and related pathways. Curr. Opin. Endocr. Metab. Res. 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Schneider, M.; Pons, J.-L.; Labesse, G.; Bourguet, W. In Silico Predictions of Endocrine Disruptors Properties. Endocrinology 2019, 160, 2709–2716. [Google Scholar] [CrossRef]
- Sun, L.; Yang, H.; Cai, Y.; Li, W.; Liu, G.; Tang, Y. In Silico Prediction of Endocrine Disrupting Chemicals Using Single-Label and Multilabel Models. J. Chem. Inf. Model. 2018, 59, 973–982. [Google Scholar] [CrossRef]
- Kolšek, K.; Mavri, J.; Dolenc, M.S.; Gobec, S.; Turk, S. Endocrine Disruptome—An Open Source Prediction Tool for Assessing Endocrine Disruption Potential through Nuclear Receptor Binding. J. Chem. Inf. Model. 2014, 54, 1254–1267. [Google Scholar] [CrossRef]
- Vedani, A.; Dobler, M.; Smieško, M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012, 261, 142–153. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Mansouri, K.; Abdelaziz, A.; Rybacka, A.; Roncaglioni, A.; Tropsha, A.; Varnek, A.; Zakharov, A.; Worth, A.; Richard, A.M.; Grulke, C.M.; et al. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect. 2016, 124, 1023–1033. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Réau, M.; Lagarde, N.; Zagury, J.-F.; Montes, M. Hits Discovery on the Androgen Receptor: In Silico Approaches to Identify Agonist Compounds. Cells 2019, 8, 1431. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ben Nasr, N.; Guillemain, H.; Lagarde, N.; Zagury, J.-F.; Montes, M. Multiple Structures for Virtual Ligand Screening: Defining Binding Site Properties-Based Criteria to Optimize the Selection of the Query. J. Chem. Inf. Model. 2013, 53, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble Docking into Multiple Crystallographically Derived Protein Structures: An Evaluation Based on the Statistical Analysis of Enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.-F.; Montes, M. Screening Explorer–An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Empereur-Mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.-F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Chemin. 2015, 7, 1–17. [Google Scholar] [CrossRef]
- Gheyouche, E.; Launay, R.; Lethiec, J.; Labeeuw, A.; Roze, C.; Amossé, A.; Téletchéa, S. DockNmine, a Web Portal to Assemble and Analyse Virtual and Experimental Interaction Data. Int. J. Mol. Sci. 2019, 20, 5062. [Google Scholar] [CrossRef]
- Danishuddin; Madhukar, G.; Malik, M.; Subbarao, N. Development and rigorous validation of antimalarial predictive models using machine learning approaches. SAR QSAR Environ. Res. 2019, 30, 543–560. [Google Scholar] [CrossRef]
- Klingspohn, W.; Mathea, M.; Ter Laak, A.; Heinrich, N.; Baumann, K. Efficiency of different measures for defining the applicability domain of classification models. J. Cheminf. 2017, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Lozach, O.; Zareifi, D.; Alexopoulos, L.; Meijer, L.; Gorgoulis, V.G.; Mikros, E. Combined Virtual and Experimental Screening for CK1 Inhibitors Identifies a Modulator of p53 and Reveals Important Aspects of in Silico Screening Performance. Int. J. Mol. Sci. 2017, 18, 2102. [Google Scholar] [CrossRef]
- Furlan, V.; Konc, J.; Bren, U. Inverse Molecular Docking as a Novel Approach to Study Anticarcinogenic and Anti-Neuroinflammatory Effects of Curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef] [PubMed]
- Réau, M.; Langenfeld, F.; Zagury, J.-F.; Lagarde, N.; Montes, M. Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef]
- Onnis, V.; Kinsella, G.K.; Carta, G.; Fayne, D.; Lloyd, D.G. Rational ligand-based virtual screening and structure–activity relationship studies in the ligand-binding domain of the glucocorticoid receptor-α. Futur. Med. Chem. 2009, 1, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.O.; Tarairah, M.; Zalloum, H.; Abu-Sheikha, G. Pharmacophore and QSAR modeling of estrogen receptor β ligands and subsequent validation and in silico search for new hits. J. Mol. Graph. Model. 2010, 28, 383–400. [Google Scholar] [CrossRef]
- Verma, N.; Chouhan, U. Chemometric Modelling of PPAR-α and PPAR-γ Dual Agonists for the Treatment of Type-2 Diabetes. Curr. Sci. 2016, 111, 356. [Google Scholar] [CrossRef]
- Lagarde, N.; Delahaye, S.; Zagury, J.-F.; Montes, M. Discriminating agonist and antagonist ligands of the nuclear receptors using 3D-pharmacophores. J. Cheminf. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kumar, V.; Kundu, B.; Bhattacharya, D.; Preethy, N.; Reddy, M.P.; Talukdar, A. Ligand-based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors. Comput. Struct. Biotechnol. J. 2019, 17, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Vittorio, S.; Seidel, T.; Germanò, M.P.; Gitto, R.; Ielo, L.; Garon, A.; Rapisarda, A.; Pace, V.; Langer, T.; De Luca, L. A Combination of Pharmacophore and Docking-based Virtual Screening to Discover new Tyrosinase Inhibitors. Mol. Inform. 2020, 39, e1900054. [Google Scholar] [CrossRef]
- Li, P.; Peng, J.; Zhou, Y.; Li, Y.; Liu, X.; Wang, L.; Zuo, Z. Discovery of FIXa inhibitors by combination of pharmacophore modeling, molecular docking, and 3D-QSAR modeling. J. Recept. Signal Transduct. 2018, 38, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Shi, Y.; Le, G. Statistical methods and molecular docking for the prediction of thyroid hormone receptor subtype binding affinity and selectivity. Struct. Chem. 2017, 28, 833–847. [Google Scholar] [CrossRef]
- Lu, S.-H.; Wu, J.W.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Epoliti, R.; Eisayev, O.; Efarag, S.; Etropsha, A. QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci. 2016, 4, 4. [Google Scholar] [CrossRef]
- Russo, D.P.; Zorn, K.M.; Clark, A.M.; Zhu, H.; Ekins, S. Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol. Pharm. 2018, 15, 4361–4370. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Chen, J.-Y.; Hor, C.-Y.; Chuang, Y.-C.; Yang, C.-B.; Yang, C.-N. Computational Study of Estrogen Receptor-Alpha Antagonist with Three-Dimensional Quantitative Structure-Activity Relationship, Support Vector Regression, and Linear Regression Methods. Available online: https://www.hindawi.com/journals/ijmc/2013/743139/ (accessed on 17 November 2020).
- Bhhatarai, B.; Wilson, D.M.; Price, P.S.; Marty, S.; Parks, A.K.; Carney, E. Evaluation of OASIS QSAR Models Using ToxCast™ in Vitro Estrogen and Androgen Receptor Binding Data and Application in an Integrated Endocrine Screening Approach. Environ. Health Perspect. 2016, 124, 1453–1461. [Google Scholar] [CrossRef]
- Rybacka, A.; Rudén, C.; Tetko, I.V.; Andersson, P.L. Identifying potential endocrine disruptors among industrial chemicals and their metabolites—Development and evaluation of in silico tools. Chemosphere 2015, 139, 372–378. [Google Scholar] [CrossRef]
- Zorn, K.M.; Foil, D.H.; Lane, T.R.; Russo, D.P.; Hillwalker, W.; Feifarek, D.J.; Jones, F.; Klaren, W.D.; Brinkman, A.M.; Ekins, S. Machine Learning Models for Estrogen Receptor Bioactivity and Endocrine Disruption Prediction. Environ. Sci. Technol. 2020, 54, 12202–12213. [Google Scholar] [CrossRef]
- Trisciuzzi, D.; Alberga, D.; Mansouri, K.; Judson, R.S.; Cellamare, S.; Catto, M.; Carotti, A.; Benfenati, E.; Novellino, E.; Mangiatordi, G.F.; et al. Docking-based classification models for exploratory toxicology studies on high-quality estrogenic experimental data. Futur. Med. Chem. 2015, 7, 1921–1936. [Google Scholar] [CrossRef]
- Zhang, L.; Sedykh, A.; Tripathi, A.; Zhu, H.; Afantitis, A.; Mouchlis, V.D.; Melagraki, G.; Rusyn, I.; Tropsha, A. Identification of putative estrogen receptor-mediated endocrine disrupting chemicals using QSAR- and structure-based virtual screening approaches. Toxicol. Appl. Pharmacol. 2013, 272, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.W.; Zhang, W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Competitive molecular docking approach for predicting estrogen receptor subtype α agonists and antagonists. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Wang, X.; Hong, H.; Benfenati, E.; Giesy, J.P.; Gini, G.C.; Kusko, R.; Zhang, X.; Yu, H.; Shi, W. Structures of Endocrine-Disrupting Chemicals Determine Binding to and Activation of the Estrogen Receptor α and Androgen Receptor. Environ. Sci. Technol. 2020, 54, 11424–11433. [Google Scholar] [CrossRef] [PubMed]
- Balaguer, P.; Delfosse, V.; Grimaldi, M.; Bourguet, W. Structural and functional evidences for the interactions between nuclear hormone receptors and endocrine disruptors at low doses. Comptes Rendus Biol. 2017, 340, 414–420. [Google Scholar] [CrossRef]
- Wassermann, A.M.; Bajorath, J.; Binding, D.B. ChEMBL: Online compound databases for drug discovery. Expert Opin. Drug Discov. 2011, 6, 683–687. [Google Scholar] [CrossRef]
- Valsecchi, C.; Grisoni, F.; Motta, S.; Bonati, L.; Ballabio, D. NURA: A curated dataset of nuclear receptor modulators. Toxicol. Appl. Pharmacol. 2020, 407, 115244. [Google Scholar] [CrossRef]
- Lagarde, N.; Delahaye, S.; Jérémie, A.; Ben Nasr, N.; Guillemain, H.; Empereur-Mot, C.; Laville, V.; Labib, T.; Réau, M.; Langenfeld, F.; et al. Discriminating Agonist from Antagonist Ligands of the Nuclear Receptors Using Different Chemoinformatics Approaches. Mol. Inform. 2017, 36, 1700020. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Grulke, C.M.; Edwards, J.; McEachran, A.D.; Mansouri, K.; Baker, N.C.; Patlewicz, G.; Shah, I.; Wambaugh, J.F.; Judson, R.S.; et al. The CompTox Chemistry Dashboard: A community data resource for environmental chemistry. J. Chemin. 2017, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.M.; Judson, R.S.; Houck, K.A.; Grulke, C.M.; Volarath, P.; Thillainadarajah, I.; Yang, C.; Rathman, J.F.; Martin, M.T.; Wambaugh, J.F.; et al. ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem. Res. Toxicol. 2016, 29, 1225–1251. [Google Scholar] [CrossRef]
- Réau, M.; Lagarde, N.; Zagury, J.-F.; Montes, M. Nuclear Receptors Database Including Negative Data (NR-DBIND): A Database Dedicated to Nuclear Receptors Binding Data Including Negative Data and Pharmacological Profile. J. Med. Chem. 2019, 62, 2894–2904. [Google Scholar] [CrossRef]
- Shen, J.; Xu, L.; Fang, H.; Richard, A.M.; Bray, J.D.; Judson, R.S.; Zhou, G.; Colatsky, T.J.; Aungst, J.L.; Teng, C.; et al. EADB: An Estrogenic Activity Database for Assessing Potential Endocrine Activity. Toxicol. Sci. 2013, 135, 277–291. [Google Scholar] [CrossRef]
- Ng, H.W.; Perkins, R.; Tong, W.; Hong, H. Versatility or Promiscuity: The Estrogen Receptors, Control of Ligand Selectivity and an Update on Subtype Selective Ligands. Int. J. Environ. Res. Public Health 2014, 11, 8709–8742. [Google Scholar] [CrossRef]
- ChemAxon—Software Solutions and Services for Chemistry & Biology. Available online: https://chemaxon.com/ (accessed on 27 July 2020).
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef]
- RDKit. Available online: https://www.rdkit.org/ (accessed on 27 July 2020).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Bank, R.P.D. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 6 May 2020).
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In Ant Colony Optimization and Swarm Intelligence; Dorigo, M., Gambardella, L.M., Birattari, M., Martinoli, A., Poli, R., Stützle, T., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2006; Volume 4150, pp. 247–258. ISBN 978-3-540-38482-3. [Google Scholar]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Pret-tenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Kainrad, T.; Hunold, S.; Seidel, T.; Langer, T. LigandScout Remote: A New User-Friendly Interface for HPC and Cloud Resources. J. Chem. Inf. Model. 2018, 59, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Docking Approach | Best Performances | Min AUC | Mean AUC | SD | |
|---|---|---|---|---|---|---|
| AUC | PDB | |||||
| smina-dkoes | Single | 0.708 | [1qku] | 0.700 | 0.704 | 0.003 |
| Ensemble of 2 | 0.709 | [2yja-1qku] | 0.702 | 0.703 | 0.003 | |
| Ensemble of 3 | 0.710 | [2yja-1qku-1g50] | 0.704 | 0.702 | 0.003 | |
| smina-vina | Single structure | 0.699 | [1a52] | 0.643 | 0.676 | 0.02 |
| Ensemble of 2 | 0.696 | [1xp9-1a52] | 0.642 | 0.67 | 0.017 | |
| Ensemble of 3 | 0.695 | [1xp9-1xp1-1a52] | 0.642 | 0.667 | 0.014 | |
| smina-vinardo | Single structure | 0.68 | [1a52] | 0.686 | 0.704 | 0.018 |
| Ensemble of 2 | 0.676 | [1xp9-1a52] | 0.619 | 0.650 | 0.019 | |
| Ensemble of 3 | 0.673 | [1xp9-1xp1-1a52] | 0.618 | 0.644 | 0.018 | |
| smina-ad4 | Single structure | 0.656 | [1a52] | 0.613 | 0.639 | 0.0154 |
| Ensemble of 2 | 0.654 | [1x7e-1a52] | 0.618 | 0.641 | 0.009 | |
| Ensemble of 3 | 0.650 | [1x7e-1qku-1a52] | 0.623 | 0.640 | 0.007 | |
| PLANTS | Single structure | 0.659 | [1x7e] | 0.598 | 0.634 | 0.019 |
| Ensemble of 2 | 0.660 | [1x7e-1a52] | 0.647 | 0.62 | 0 | |
| Ensemble of 3 | 0.659 | [1x7e-1qku-1a52] | 0.620 | 0.642 | 0.009 | |
| Surflex-dock | Single structure | 0.604 | [1a52] | 0.547 | 0.576 | 0.027 |
| Ensemble of 2 | 0.616 | [1xp1-1x7e] | 0.556 | 0.594 | 0.020 | |
| Ensemble of 3 | 0.623 | [1xp1-1x7e-1a52] | 0.562 | 0.605 | 0.015 | |
| Docking Approach | Performances | Se = 0.25 | Se = 0.5 | Se = 0.75 | |
|---|---|---|---|---|---|
| smina_dkoes | Single | P(active) | 0.137 | 0.094 | 0.094 |
| (1qku) | TH | −7 | −6 | −6 | |
| Sp | 0.918 | 0.766 | 0.601 | ||
| EF | 1.9 | 1.65 | 1.65 | ||
| PPV | 56/237 | 111/631 | 167/1052 | ||
| Ensemble de 2 | P(active) | 0.134 | 0.094 | 0.094 | |
| (2yja-1qku) | TH | −7 | −6 | −6 | |
| Sp | 0.916 | 0.759 | 0.597 | ||
| EF | 1.89 | 1.63 | 1.63 | ||
| PPV | 56/242 | 111/645 | 167/1061 | ||
| Ensemble de 3 | P(active) | 0.137 | 0.13 | 0.091 | |
| (2yja-1qku-1g50) | TH | −8 | −7 | −6 | |
| Sp | 0.915 | 0.777 | 0.599 | ||
| EF | 2.37 | 1.9 | 1.59 | ||
| PPV | 56/244 | 111/605 | 167/1057 | ||
| PLANTS | Single | P(active) | 0.127 | 0.103 | 0.081 |
| (1x7e) | TH | −79 | −72 | −64 | |
| Sp | 0.876 | 0.723 | 0.501 | ||
| EF | 1.9 | 1.69 | 1.42 | ||
| PPV | 55/328 | 110/719 | 165/1261 | ||
| Ensemble of 2 | P(active) | 0.123 | 0.097 | 0.08 | |
| (1x7e-1a52) | TH | −82 | −73 | −66 | |
| Sp | 0.86 | 0.707 | 0.49 | ||
| EF | 1.69 | 1.58 | 1.42 | ||
| PPV | 55/362 | 110/753 | 165/1287 | ||
| Ensemble of 3 | P(active) | 0.122 | 0.096 | 0.079 | |
| (1x7e-1a52-1qku) | TH | −82 | −73 | −66 | |
| Sp | 0.857 | 0.701 | 0.493 | ||
| EF | 1.65 | 1.6 | 1.41 | ||
| PPV | 55/369 | 110/767 | 165/1279 |
| Scoring Threshold (TH) | Performances | EPA | Estrogenic Activity DataBase (EADB) | Nuclear Receptors DataBase Including Negative Data (NR-DBIND) |
|---|---|---|---|---|
| TH = −7 | Se | 0.79 | 0.48 | 0.93 |
| Sp | 0.55 | 0.58 | 0.03 | |
| PPV | 176/2442 | 63/232 | 513/732 | |
| TH = −6 | Se | 0.46 | 0.77 | 0.99 |
| Sp | 0.78 | 0.198 | 0.001 | |
| PPV | 103/2442 | 101/232 | 553/732 |
| EPA Database | EADB | NR-DBIND | |||
|---|---|---|---|---|---|
| Performances | Train Set | Test Set | Validation Set | Validation Set | |
| LB pharmacophores | Se (B/total_B) | 0.305 (51/167) | 0.232 (13/56) | ||
| Sp (NB/total_NB) | 0.973 (45/1664) | 0.960 (22/555) | |||
| SB pharmacophores | Se (B/total_B) | 0.251 (42/167) | 0.232 (13/56) | ||
| Sp (NB/total_NB) | 0.990 (16/1664) | 0.987 (7/555) | |||
| SBLB pharmacophores | Se (B/total_B) | 0.371 (62/1664) | 0.321 (18/56) | 0.557 (73/131) | 0.819 (458/554) |
| Sp (NB/total_NB) | 0.968 (53/167) | 0.595 (25/555) | 0.871 (13/101) | 0.629 (66/178) | |
| TH | −7 | −6 | |||||
|---|---|---|---|---|---|---|---|
| Se | Sp | PPV | Se | Sp | PPV | ||
| Consensus protocol | EPA database | 0.56 | 0.76 | 124/652 | 0.81 | 0.54 | 180/1205 |
| EADB | 0.832 | 0.495 | 109/160 | 0.931 | 0.158 | 122/207 | |
| NR-DBIND | 0.986 | 0.029 | 546/719 | 1.0 | 0.005 | 554/731 | |
| Hierarchical protocol | EPA database | 0.25 | 0.99 | 55/84 | 0.32 | 0.98 | 72/117 |
| EADB | 0.206 | 0.960 | 27/31 | 0.370 | 0.911 | 52/61 | |
| NR-DBIND | 0.756 | 0.635 | 419/484 | 0.814 | 0.635 | 451/516 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sellami, A.; Montes, M.; Lagarde, N. Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods. Int. J. Mol. Sci. 2021, 22, 2846. https://doi.org/10.3390/ijms22062846
Sellami A, Montes M, Lagarde N. Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods. International Journal of Molecular Sciences. 2021; 22(6):2846. https://doi.org/10.3390/ijms22062846
Chicago/Turabian StyleSellami, Asma, Matthieu Montes, and Nathalie Lagarde. 2021. "Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods" International Journal of Molecular Sciences 22, no. 6: 2846. https://doi.org/10.3390/ijms22062846
APA StyleSellami, A., Montes, M., & Lagarde, N. (2021). Predicting Potential Endocrine Disrupting Chemicals Binding to Estrogen Receptor α (ERα) Using a Pipeline Combining Structure-Based and Ligand-Based in Silico Methods. International Journal of Molecular Sciences, 22(6), 2846. https://doi.org/10.3390/ijms22062846