
Role of ADAM10 and ADAM17 in Regulating CD137 Function



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
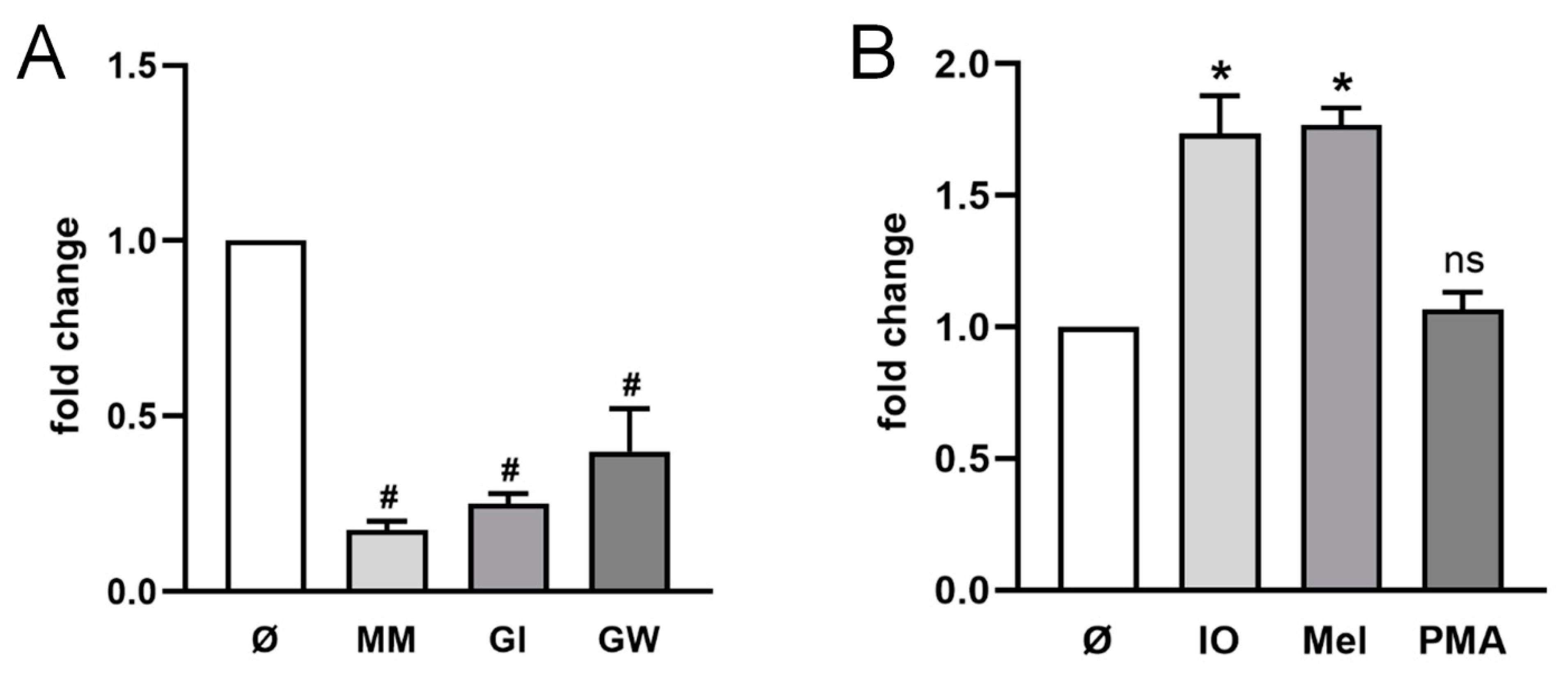
2.1. ADAM10 Is the Major Sheddase of CD137 in HT29 Cells
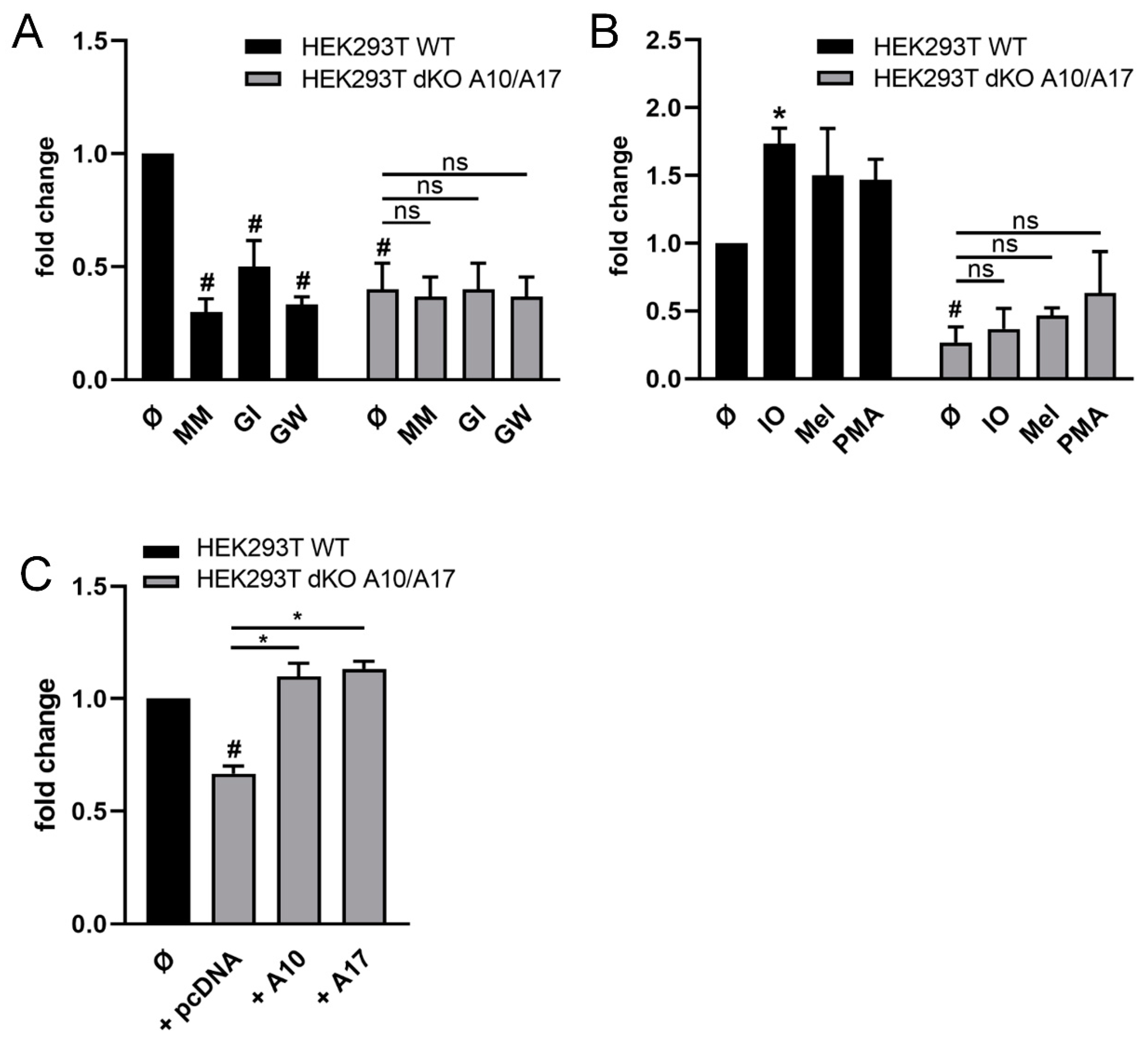
2.2. ADAM10 and ADAM17 Can Release CD137 in HEK Cells
2.3. Ionomycin-Induced Shedding of CD137 Is PS Dependent
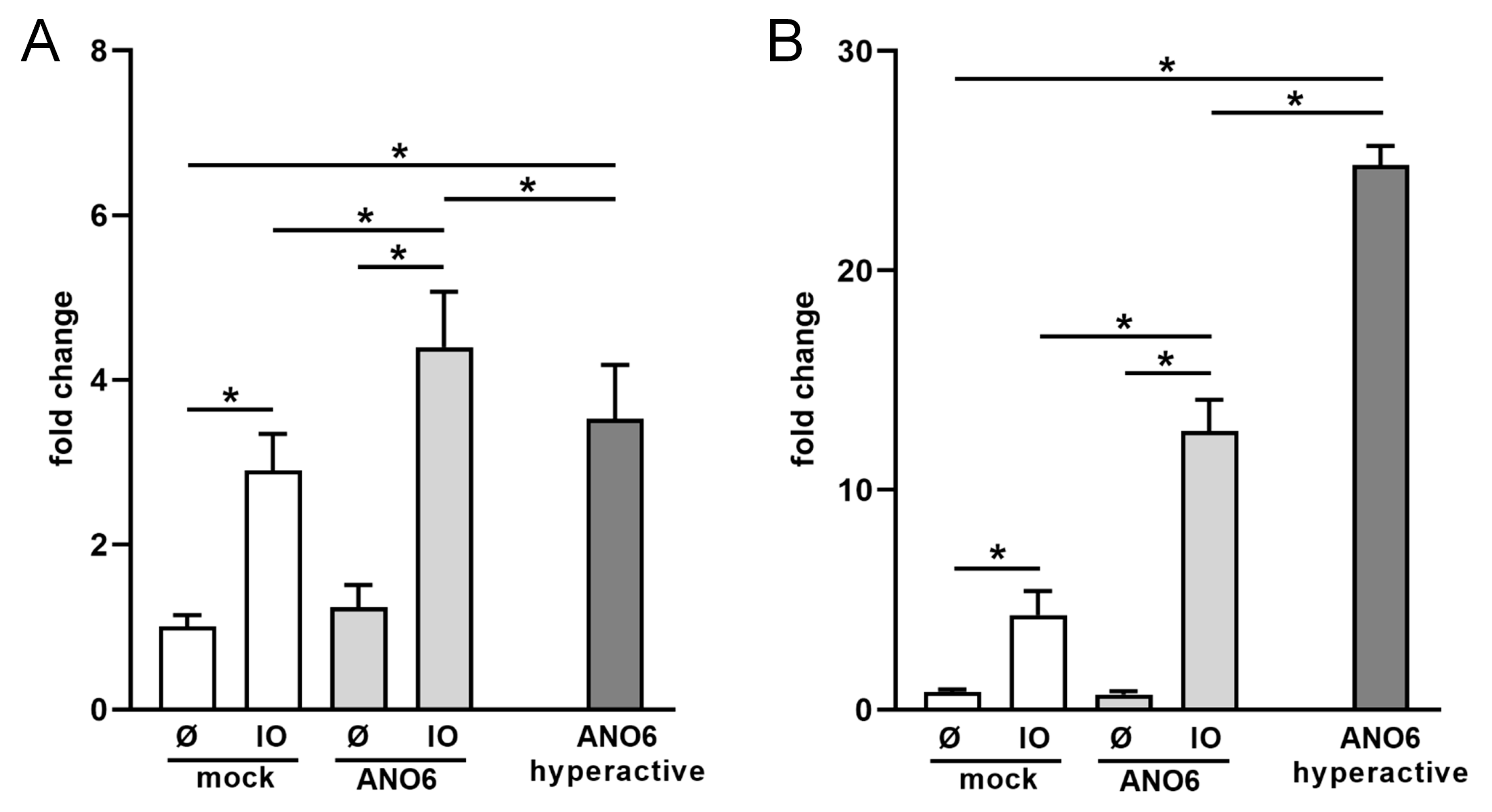
2.4. Anoctamin-6 Modulates CD137 Release
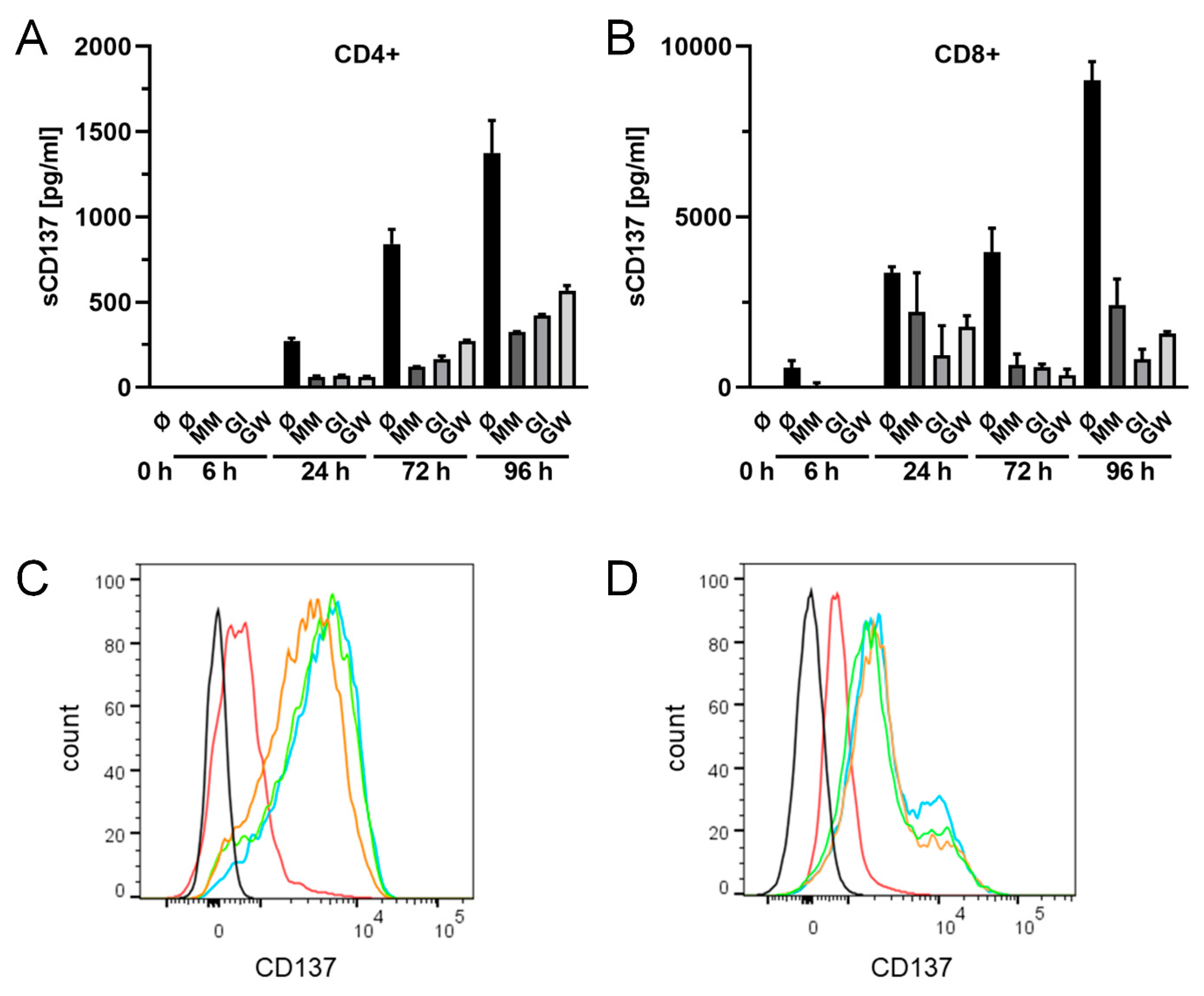
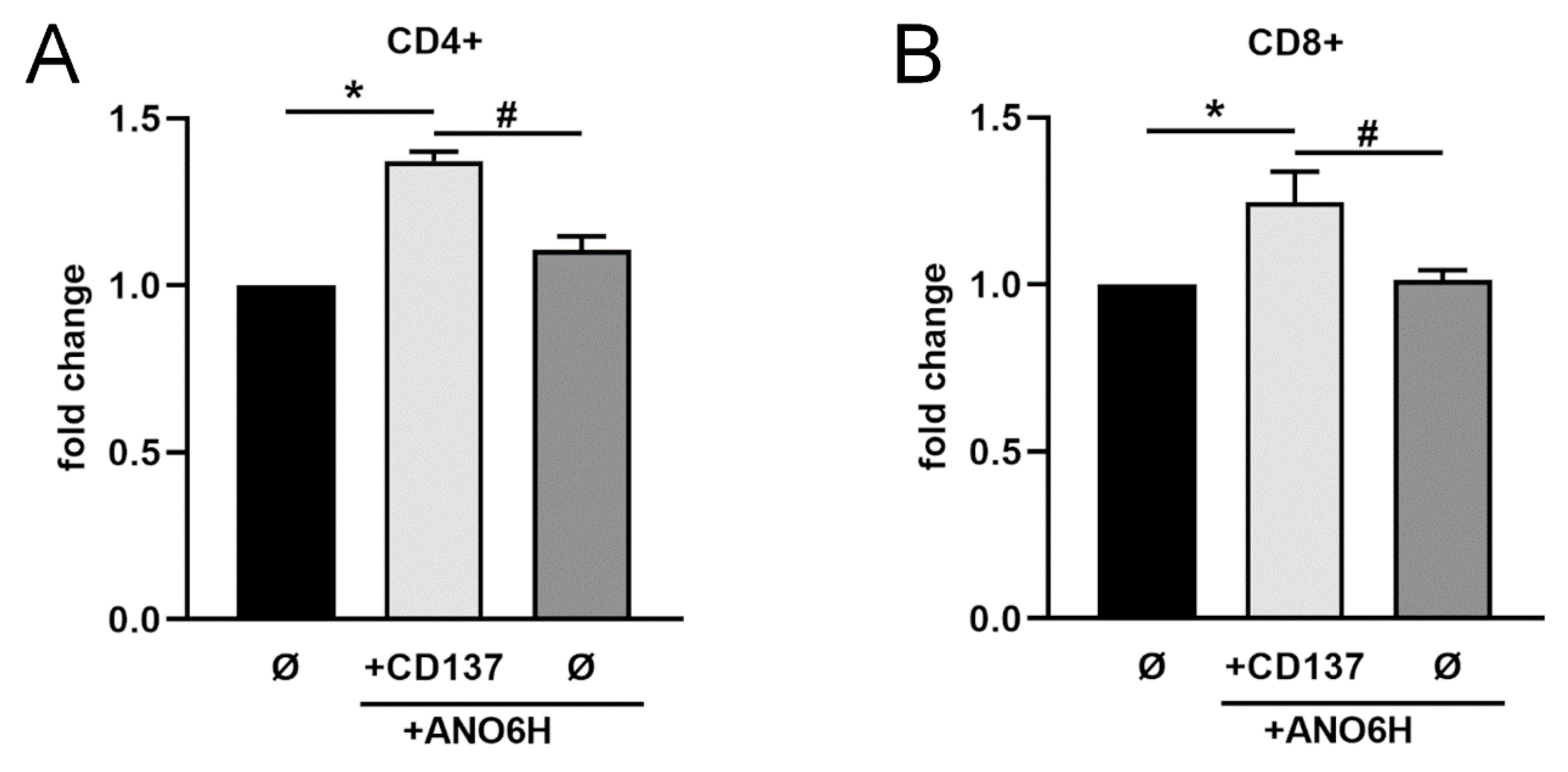
2.5. Release of Endogenously Expressed CD137 in T Cells
2.6. Shed sCD137 Induces Proliferation of Activated T Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. T Cell Isolation and FACS Analysis
4.4. Expression Vectors and Transfection
4.5. Enzyme-Linked Immunosorbent Assay
4.6. Proliferation Assay
4.7. Semi-Quantitative RT-PCR
4.8. Automated Western
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, B.S.; Weissman, S.M. cDNA sequences of two inducible T-cell genes. Proc. Natl. Acad. Sci. USA 1989, 86, 1963–1967. [Google Scholar] [CrossRef]
- Schwarz, H.; Tuckwell, J.; Lotz, M. A receptor induced by lymphocyte activation (ILA): A new member of the human nerve-growth-factor/tumor-necrosis-factor receptor family. Gene 1993, 134, 295–298. [Google Scholar] [CrossRef]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, Á.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2020, 4. [Google Scholar] [CrossRef]
- Wong, H.Y.; Schwarz, H. CD137/CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J. Autoimmun. 2020, 112. [Google Scholar] [CrossRef]
- Choi, B.K.; Lee, H.W. The Murine CD137/CD137 Ligand Signalosome: A Signal Platform Generating Signal Complexity. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Langstein, J.; Michel, J.; Fritsche, J.; Kreutz, M.; Andreesen, R.; Schwarz, H. CD137 (ILA/4-1BB), a member of the TNF receptor family, induces monocyte activation via bidirectional signaling. J. Immunol. 1998, 160, 2488–2494. [Google Scholar] [PubMed]
- Shao, Z.; Schwarz, H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J. Leukoc. Biol. 2011, 89, 21–29. [Google Scholar] [CrossRef]
- Goodwin, R.G.; Din, W.S.; Davis-Smith, T.; Anderson, D.M.; Gimpel, S.D.; Sato, T.A.; Maliszewski, C.R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: A member of an emerging family of cytokines with homology to tumor necrosis factor. Eur. J. Immunol. 1993, 23, 2631–2641. [Google Scholar] [CrossRef] [PubMed]
- Alderson, M.R.; Smith, C.A.; Tough, T.W.; Davis-Smith, T.; Armitage, R.J.; Falk, B.; Roux, E.; Baker, E.; Sutherland, G.R.; Din, W.S.; et al. Molecular and biological characterization of human 4-1BB and its ligands. Eur. J. Immunol. 1994, 24, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Pollok, K.E.; Kim, Y.-J.; Hurtado, J.; Zhou, Z.; Kim, K.K.; Kwon, B.S. 4-1BB T-cell antigen binds to mature B cells and macrophages, and costimulates anti-μ-primed splenic B cells. Eur. J. Immunol. 1994, 24, 367–374. [Google Scholar] [CrossRef]
- Wang, S.; Chen, L. Immunobiology of Cancer Therapies Targeting CD137 and B7-H1/PD-1 Cosignal Pathways. Curr. Top. Microbiol. Immunol. 2010, 344, 245–267. [Google Scholar] [CrossRef]
- Thum, E.; Zhe, S.; Schwarz, H. CD137, implications in immunity and potential for therapy. Front. Biosci. 2009, 14, 4173–4188. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Furtner, M.; Straub, R.H.; Krüger, S.; Schwarz, H. Levels of soluble CD137 are enhanced in sera of leukemia and lymphoma patients and are strongly associated with chronic lymphocytic leukemia. Leukemia 2005, 19, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, P.; Zhang, Q.; Wang, X.; Li, J.; Ma, C.; Sun, W.; Zhang, L. Analysis of CD137 and CD137L expression in human primary tumor tissues. Croat. Med. J. 2008, 49, 192–200. [Google Scholar] [CrossRef]
- Grimmig, T.; Gasser, M.; Moench, R.; Zhu, L.J.; Nawalaniec, K.; Callies, S.; Wagner, M.; Polat, B.; Mothi, S.S.; Luo, Y.; et al. Expression of Tumor-mediated CD137 ligand in human colon cancer indicates dual signaling effects. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef]
- Glorieux, C.; Huang, P. Regulation of CD137 expression through K-Ras signaling in pancreatic cancer cells. Cancer Commun. 2019, 39, 41. [Google Scholar] [CrossRef]
- Jin, J.; Jung, I.H.; Moon, S.H.; Jeon, S.; Jeong, S.J.; Sonn, S.K.; Seo, S.; Lee, M.N.; Song, E.J.; Kweon, H.Y.; et al. CD137 Signaling Regulates Acute Colitis via RALDH2-Expressing CD11b−CD103+ DCs. Cell Rep. 2020, 30, 4124–4136.e5. [Google Scholar] [CrossRef]
- Söderström, L.; Tarnawski, L.; Olofsson, P.S. CD137: A checkpoint regulator involved in atherosclerosis. Atherosclerosis 2018, 272, 66–72. [Google Scholar] [CrossRef]
- He, Y.; Ao, D.H.; Li, X.Q.; Zhong, S.S.; Rong, A.; Wang, Y.Y.; Xiang, Y.J.; Xu, B.L.; Yang, T.T.; Gao, X.G.; et al. Increased Soluble CD137 Levels and CD4+ T-Cell-Associated Expression of CD137 in Acute Atherothrombotic Stroke. Clin. Transl. Sci. 2018, 11, 428–434. [Google Scholar] [CrossRef]
- Michel, J.; Langstein, J.; Hofstädter, F.; Schwarz, H. A soluble form of CD137 (ILA/4-1BB), a member of the TNF receptor family, is released by activated lymphocytes and is detectable in sera of patients with rheumatoid arthritis. Eur. J. Immunol. 1998, 28, 290–295. [Google Scholar] [CrossRef]
- Dimberg, J.; Hugander, A.; Wågsäter, D. Expression of CD137 and CD137 ligand in colorectal cancer patients. Oncol. Rep. 2006, 15, 1197–1200. [Google Scholar] [CrossRef][Green Version]
- Yan, J.; Wang, C.; Chen, R.; Yang, H. Clinical implications of elevated serum soluble CD137 levels in patients with acute coronary syndrome. Clinics 2013, 68, 193–198. [Google Scholar] [CrossRef]
- Dongming, L.; Zuxun, L.; Liangjie, X.; Biao, W.; Ping, Y. Enhanced levels of soluble and membrane-bound CD137 levels in patients with acute coronary syndromes. Clin. Chim. Acta 2010, 411, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.W.; Choi, S.W.; Choi, J.I.L.; Kwon, B.S. Serum concentrations of soluble 4-1BB, and 4-1BB ligand correlated with the disease severity in rheumatoid arthritis. Exp. Mol. Med. 2004, 36, 13–22. [Google Scholar] [CrossRef]
- Shao, Z.; Sun, F.; Koh, D.R.; Schwarz, H. Characterisation of soluble murine CD137 and its association with systemic lupus. Mol. Immunol. 2008, 45, 3990–3999. [Google Scholar] [CrossRef]
- Luu, K.; Shao, Z.; Schwarz, H. The relevance of soluble CD137 in the regulation of immune responses and for immunotherapeutic intervention. J. Leukoc. Biol. 2020, 107, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Slack, J.L.; Davis, R.; Cerretti, D.P.; Kozlosky, C.J.; Blanton, R.A.; Shows, D.; Peschon, J.J.; Black, R.A. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 2000, 275, 14608–14614. [Google Scholar] [CrossRef]
- Bell, J.H.; Herrera, A.H.; Li, Y.; Walcheck, B. Role of ADAM17 in the ectodomain shedding of TNF- and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 2007, 82, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Taraban, V.Y.; Rowley, T.F.; O’Brien, L.; Claude Chan, H.T.; Haswell, L.E.; Green, M.H.A.; Tutt, A.L.; Glennie, M.J.; Al-Shamkhani, A. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur. J. Immunol. 2002, 32, 3617–3627. [Google Scholar] [CrossRef]
- Kagawa, K.; Nakano, A.; Miki, H.; Oda, A.; Amou, H.; Takeuchi, K.; Nakamura, S.; Harada, T.; Fujii, S.; Yata, K.; et al. Inhibition of tace activity enhances the susceptibility of myeloma cells to TRAIL. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Hakozaki, A.; Yoda, M.; Tohmonda, T.; Furukawa, M.; Hikata, T.; Uchikawa, S.; Takaishi, H.; Matsumoto, M.; Chiba, K.; Horiuchi, K.; et al. Receptor Activator of NF-κB (RANK) Ligand Induces Ectodomain Shedding of RANK in Murine RAW264.7 Macrophages. J. Immunol. 2010, 184, 2442–2448. [Google Scholar] [CrossRef]
- Sommer, A.; Kordowski, F.; Büch, J.; Maretzky, T.; Evers, A.; Andrä, J.J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef]
- Veit, M.; Koyro, K.I.; Ahrens, B.; Bleibaum, F.; Munz, M.; Rövekamp, H.; Andrä, J.; Schreiber, R.; Kunzelmann, K.; Sommer, A.; et al. Anoctamin-6 regulates ADAM sheddase function. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Loughran, P.A.; Zhang, L.; Scott, M.J.; Billiar, T.R. Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci. Signal. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Defamie, V.; Smookler, D.S.; Di Grappa, M.A.; Horiuchi, K.; Federici, M.; Sibilia, M.; Blobel, C.P.; Khokha, R. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J. Clin. Investig. 2010, 120, 2731–2744. [Google Scholar] [CrossRef]
- Terrado, J.; Monnier, D.; Perrelet, D.; Vesin, D.; Jemelin, S.; Buurman, W.A.; Mattenberger, L.; King, B.; Kato, A.C.; Garcia, I. Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur. J. Neurosci. 2000, 12, 3443–3447. [Google Scholar] [CrossRef]
- Xia, M.; Xue, S.B.; Xu, C.S. Shedding of TNFR1 in regenerative liver can be induced with TNF α and PMA. World J. Gastroenterol. 2002, 8, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, T.; Ishii, S.; Nishimi, S.; Nishimi, A.; Oguro, N.; Seki, S.; Miura, Y.; Miwa, Y.; Oh, K.; Toyoshima, Y.; et al. A disintegrin and metalloprotease-10 is correlated with disease activity and mediates monocyte migration and adhesion in rheumatoid arthritis. Transl. Res. 2015, 166, 244–253. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Isozaki, T.; Rabquer, B.J.; Ruth, J.H.; Haines, G.K.; Koch, A.E. ADAM-10 is overexpressed in rheumatoid arthritis synovial tissue and mediates angiogenesis. Arthritis Rheum. 2013, 65, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.M.; Tharakan, A.; Martin, R.K. Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 2020, 11, 499. [Google Scholar] [CrossRef]
- Sommer, A.; Fries, A.; Cornelsen, I.; Speck, N.; Koch-Nolte, F.; Gimpl, G.; Andrä, J.; Bhakdi, S.; Reiss, K. Melittin modulates keratinocyte function through P2 receptor-dependent ADAM activation. J. Biol. Chem. 2012, 287, 23678–23689. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Bobé, P.; Reiss, K.; Horiuchi, K.; Niu, X.-D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef]
- Riethmueller, S.; Ehlers, J.C.; Lokau, J.; Düsterhöft, S.; Knittler, K.; Dombrowsky, G.; Grötzinger, J.; Rabe, B.; Rose-John, S.; Garbers, C. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Veit, M.; Ahrens, B.; Seidel, J.; Sommer, A.; Bhakdi, S.; Reiss, K. Mutagenesis of the ADAM17-phosphatidylserine-binding motif leads to embryonic lethality in mice. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Shi, J.; Heegaard, C.W.; Rasmussen, J.T.; Gilbert, G.E. Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature. Biochim. Biophys. Acta Biomembr. 2004, 1667, 82–90. [Google Scholar] [CrossRef]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Pollok, K.E.; Kim, Y.J.; Zhou, Z.; Hurtado, J.; Kim, K.K.; Pickard, R.T.; Kwon, B.S. Inducible T cell antigen 4-1BB. Analysis of expression and function. J. Immunol. 1993, 150, 771–781. [Google Scholar]
- van Oers, M.; Pals, S.; Evers, L.; van der Schoot, C.; Koopman, G.; Bonfrer, J.; Hintzen, R.; Kr von dem Borne, A.; van Lier, R. Expression and Release of CD27 in Human B-Cell Malignancies. Blood 1993, 82, 3430–3436. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Tesselaar, K.; Van Oers, M.H.J.; Van Lier, R.A.W. Control of lymphocyte function through CD27-CD70 interactions. Semin. Immunol. 1998, 10, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jochems, C.; Anderson, A.M.; Talaie, T.; Jales, A.; Madan, R.A.; Hodge, J.W.; Tsang, K.Y.; Liewehr, D.J.; Steinberg, S.M.; et al. Soluble CD27-Pool in Humans May Contribute to T Cell Activation and Tumor Immunity. J. Immunol. 2013, 190, 6250–6258. [Google Scholar] [CrossRef]
- Möller-Hackbarth, K.; Dewitz, C.; Schweigert, O.; Trad, A.; Garbers, C.; Rose-John, S.; Scheller, J. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3). J. Biol. Chem. 2013, 288, 34529–34544. [Google Scholar] [CrossRef] [PubMed]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM. Cell 2017, 171, 1–15. [Google Scholar] [CrossRef]
- Tu, T.H.; Kim, C.S.; Kang, J.H.; Nam-Goong, I.S.; Nam, C.W.; Kim, E.S.; Kim, Y.I.; Choi, J.I.; Kawada, T.; Goto, T.; et al. Levels of 4-1BB transcripts and soluble 4-1BB protein are elevated in the adipose tissue of human obese subjects and are associated with inflammatory and metabolic parameters. Int. J. Obes. 2014, 38, 1075–1082. [Google Scholar] [CrossRef]
- Eun, S.-Y.; Lee, S.-W.; Xu, Y.; Croft, M. 4-1BB Ligand Signaling to T Cells Limits T Cell Activation. J. Immunol. 2015, 194, 134–141. [Google Scholar] [CrossRef]
- Ishizaka, A.; Tono-oka, T.; Matsumoto, S. Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Methods 1984, 72, 127–132. [Google Scholar] [CrossRef]
- Petty, R.D.; Sutherland, L.A.; Hunter, E.M.; Cree, I.A. Comparison of MTT and ATP-based assays for the measurement of viable cell number. J. Biolumin. Chemilumin. 1995, 10, 29–34. [Google Scholar] [CrossRef]
- Crouch, S.P.M.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic consequences of T-cell costimulation in anticancer immunity. Cancer Immunol. Res. 2019, 7, 1664–1669. [Google Scholar] [CrossRef]
- Labiano, S.; Palazón, A.; Bolaños, E.; Azpilikueta, A.; Sánchez-Paulete, A.R.; Morales-Kastresana, A.; Quetglas, J.I.; Perez-Gracia, J.L.; Gúrpide, A.; Rodriguez-Ruiz, M.; et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology 2016, 5, e1062967. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; David Becherer, J. Metalloproteinase Inhibitors for the Disintegrin-Like Metalloproteinases ADAM10 and ADAM17 that Differentially Block Constitutive and Phorbol Ester-Inducible Shedding of Cell Surface Molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Andrä, J.; Koch, M.H.J.; Bartels, R.; Brandenburg, K. Biophysical Characterization of Endotoxin Inactivation by NK-2, an Antimicrobial Peptide Derived from Mammalian NK-Lysin. Antimicrob. Agents Chemother. 2004, 48, 1593–1599. [Google Scholar] [CrossRef]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 2000, 347, 131–138. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, J.; Leitzke, S.; Ahrens, B.; Sperrhacke, M.; Bhakdi, S.; Reiss, K. Role of ADAM10 and ADAM17 in Regulating CD137 Function. Int. J. Mol. Sci. 2021, 22, 2730. https://doi.org/10.3390/ijms22052730
Seidel J, Leitzke S, Ahrens B, Sperrhacke M, Bhakdi S, Reiss K. Role of ADAM10 and ADAM17 in Regulating CD137 Function. International Journal of Molecular Sciences. 2021; 22(5):2730. https://doi.org/10.3390/ijms22052730
Chicago/Turabian StyleSeidel, Jana, Sinje Leitzke, Björn Ahrens, Maria Sperrhacke, Sucharit Bhakdi, and Karina Reiss. 2021. "Role of ADAM10 and ADAM17 in Regulating CD137 Function" International Journal of Molecular Sciences 22, no. 5: 2730. https://doi.org/10.3390/ijms22052730
APA StyleSeidel, J., Leitzke, S., Ahrens, B., Sperrhacke, M., Bhakdi, S., & Reiss, K. (2021). Role of ADAM10 and ADAM17 in Regulating CD137 Function. International Journal of Molecular Sciences, 22(5), 2730. https://doi.org/10.3390/ijms22052730