
Drosophila as a Model for Infectious Diseases



Abstract
1. Introduction
2. Innate Immune Signaling Pathways and Pathogenic Proteins that Affect Them
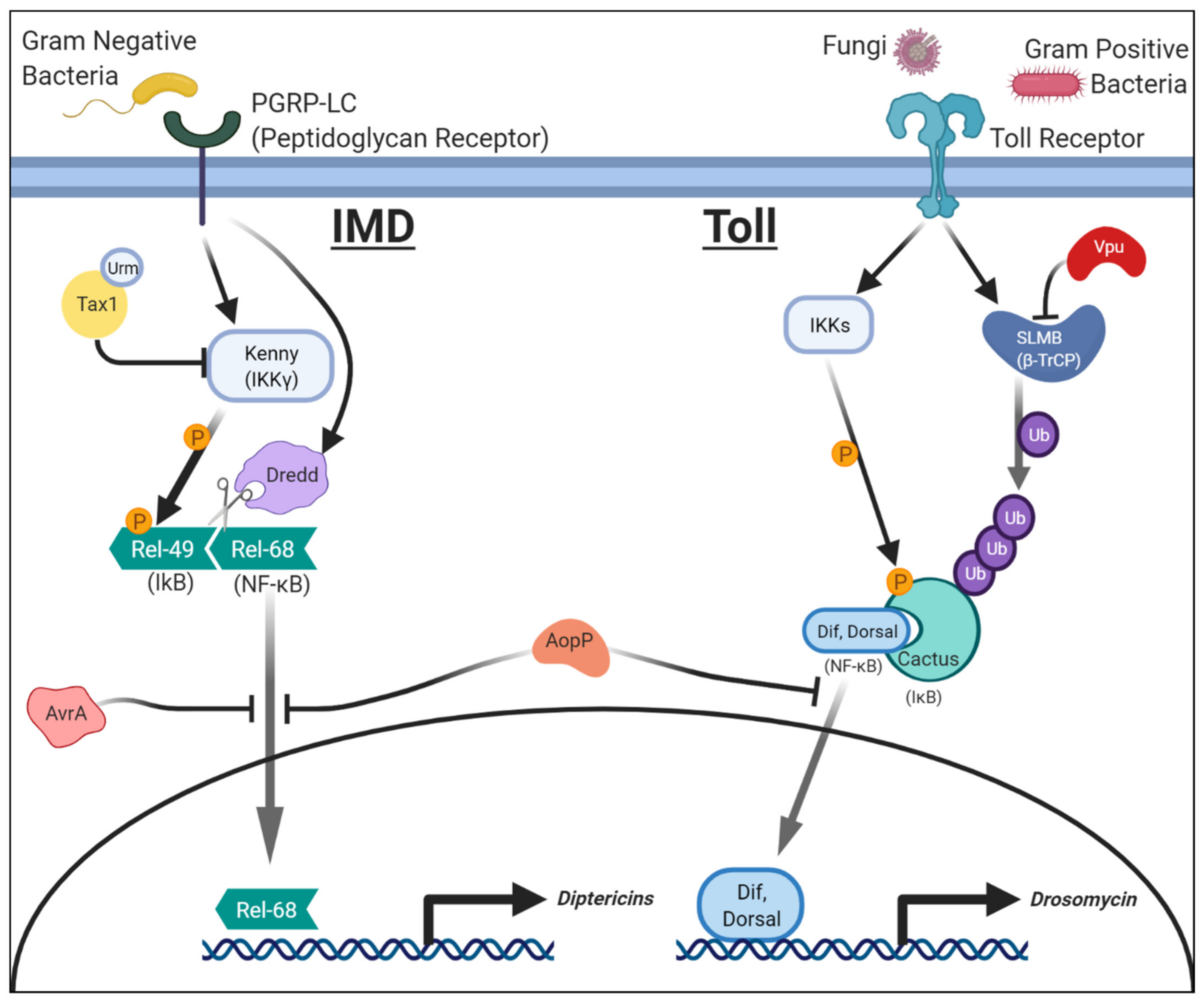
2.1. Salmonellae Enterica: AvrA and NF-κB (IMD)/JNK Signaling
2.2. Aeromonas Salmonicida: AopP and NF-κB (Toll and IMD) Signaling
2.3. HTLV-1: Tax1 in NF-κB (IMD) Signaling
2.4. HIV: Vpu in NF-κB (Toll) Signaling
2.5. HIV: Nef in JNK Signaling
3. Pathogenic Proteins that Affect Phagocytosis and Apoptosis
3.1. Pseudomonas aeruginosa: ExoS in Phagocytosis
3.2. SARS-CoV-1: 3a and M in Apoptosis
3.3. HIV: Vpu in Apoptosis through JNK Signaling
3.4. EBV: BZLF1 and BRLF1 in Apoptosis and Cell Proliferation
4. Pathogenic Proteins that Affect Fundamental Cellular Processes and Developmental Signaling Pathways
4.1. Zika Virus: NS4A in Neural Stem Cell Survival and Proliferation
4.2. Zika Virus: NS4A in JAK-STAT and Notch Signaling
4.3. Bacillus Anthracis: LF and EF in Multiple Signaling Pathways and Cell Adhesion
4.4. Vibrio Cholerae: CtxA in Notch Signaling and Cell Adhesion
4.5. HCMV: Immediate-Early Genes in Cell Adhesion
4.6. Helicobacter Pylori: CagA in Multiple Signaling Pathways, Cytoskeletal Organization and Microbiome Composition
4.7. HPV: E6 in Cell Polarity and Epithelial-to-Mesenchymal Transition
4.8. HIV: Tat in Cytoskeleton Organization and Protein Translation
4.9. SV40: Large and Small T Antigens in Mitosis
5. Drosophila Studies to Identify New Therapeutic Targets to Combat Infectious Diseases
5.1. Influenza Virus: M2 in pH Regulation
5.2. Pseudomonas aeruginosa. gshA and gshB in Bacterial Stress Resistance and Biofilm Production
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Health Statistics 2020: Monitoring Health for the SDGs, Sustainable Development Goals; WHO: Geneva, Switzerland, 2020; Volume 8. [Google Scholar]
- Fauci, A.S.; Lane, H.C.; Redfield, R.R. Covid-19—Navigating the Uncharted. N. Engl. J. Med. 2020, 1268–1269. [Google Scholar] [CrossRef]
- Wangler, M.F.; Yamamoto, S.; Chao, H.T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Hieter, P.; Boycott, K.M.; Campeau, P.M.; Bellen, H.J.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 9–27. [Google Scholar] [CrossRef]
- Bellen, H.J.; Yamamoto, S. Morgan’s Legacy: Fruit Flies and the Functional Annotation of Conserved Genes. Cell 2015, 12–14. [Google Scholar] [CrossRef]
- Wangler, M.F.; Yamamoto, S.; Bellen, H.J. Fruit Flies in Biomedical Research. Genetics 2015, 199, 639–653. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an in Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef]
- Sonoshita, M.; Cagan, R.L. Modeling Human Cancers in Drosophila. Curr. Top. Dev. Biol. 2017, 121, 287–309. [Google Scholar] [CrossRef]
- Buchon, N.; Silverman, N.; Cherry, S. Immunity in Drosophila Melanogaster-from Microbial Recognition to Whole-Organism Physiology. Nat. Rev. Immunol. 2014, 796–810. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette Spatzle/Toll/Cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Bellen, H.J.; Wangler, M.F.; Yamamoto, S. The Fruit Fly at the Interface of Diagnosis and Pathogenic Mechanisms of Rare and Common Human Diseases. Hum. Mol. Genet. 2019, R207–R214. [Google Scholar] [CrossRef] [PubMed]
- Harnish, J.M.; Deal, S.L.; Chao, H.T.; Wangler, M.F.; Yamamoto, S. In Vivo Functional Study of Disease-Associated Rare Human Variants Using Drosophila. J. Vis. Exp. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted Gene Expression as a Means of Altering Cell Fates and Generating Dominant Phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Kaya-Çopur, A.; Schnorrer, F. A Guide to Genome-Wide in Vivo RNAI Applications in Drosophila. Methods Mol. Biol. 2016, 1478, 117–143. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Strein, C.; Stricker, M.; Rauscher, B.; Heigwer, F.; Zhou, J.; Beyersdörffer, C.; Frei, J.; Hess, A.; Kern, K.; et al. A Large-Scale Resource for Tissue-Specific CRISPR Mutagenesis in Drosophila. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Zirin, J.; Hu, Y.; Liu, L.; Yang-Zhou, D.; Colbeth, R.; Yan, D.; Ewen-Campen, B.; Tao, R.; Vogt, E.; VanNest, S.; et al. Large-Scale Transgenic Drosophila Resource Collections for Loss- and Gain-of-Function Studies. Genetics 2020, 214, 755–767. [Google Scholar] [CrossRef]
- Bier, E. Drosophila, the Golden Bug, Emerges as a Tool for Human Genetics. Nat. Rev. Genet. 2005, 9–23. [Google Scholar] [CrossRef]
- Cook, K.R.; Parks, A.L.; Jacobus, L.M.; Kaufman, T.C.; Matthews, K.A. New Research Resources at the Bloomington Drosophila Stock Center. Fly 2010, 4, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.T. Drosophila Genetic Resource and Stock Center; The National Bioresource Project. Exp. Anim. 2010, 125–138. [Google Scholar] [CrossRef]
- Troha, K.; Buchon, N. Methods for the Study of Innate Immunity in Drosophila Melanogaster. Wiley Interdiscip. Rev. Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.T.; Allen, A.L.; Bardin, J.E.; Christian, M.N.; Daimon, K.; Dozier, K.D.; Hansen, C.L.; Holcomb, L.M.; Ahlander, J. Drosophila as a Genetic Model for Studying Pathogenic Human Viruses. Virology 2012, 1–5. [Google Scholar] [CrossRef]
- Panayidou, S.; Ioannidou, E.; Apidianakis, Y. Human Pathogenic Bacteria, Fungi, and Viruses in Drosophila. Virulence 2014, 5, 253–269. [Google Scholar] [CrossRef]
- Boyer, L.; Paquette, N.; Silverman, N.; Stuart, L.M. Bacterial Effectors: Learning on the Fly. Adv. Exp. Med. Biol. 2012, 710, 29–36. [Google Scholar] [CrossRef]
- Neyen, C.; Bretscher, A.J.; Binggeli, O.; Lemaitre, B. Methods to Study Drosophila Immunity. Methods 2014, 68, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; Sanchez-Garcia, J.; Rincon-Limas, D.E. Drosophila Models of Prionopathies: Insight into Prion Protein Function, Transmission, and Neurotoxicity. Curr. Opin. Genet. Dev. 2017, 141–148. [Google Scholar] [CrossRef]
- Tzelepis, I.; Kapsetaki, S.E.; Panayidou, S.; Apidianakis, Y. Drosophila Melanogaster: A First Step and a Stepping-Stone to Anti-Infectives. Curr. Opin. Pharmacol. 2013, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-ΚB in Immunobiology. Cell Res. 2011, 223–244. [Google Scholar] [CrossRef]
- Hetru, C.; Hoffmann, J.A. NF-KappaB in the Immune Response of Drosophila. Cold Spring Harbor Perspect. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Park, A.J.; Okhovat, J.P.; Kim, J. Antimicrobial Peptides. In Clinical and Basic Immunodermatology, 2nd ed.; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Tanji, T.; Yun, E.Y.; Ip, Y.T. Heterodimers of NF-ΚB Transcription Factors DIF and Relish Regulate Antimicrobial Peptide Genes in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 14715–14720. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-ΚB P65 and Strategies for Therapeutic Manipulation. J. Inflamm. Res. 2018, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Stöven, S.; Silverman, N.; Junell, A.; Hedengren-Olcott, M.; Erturk, D.; Engström, Y.; Maniatis, T.; Hultmark, D. Caspase-Mediated Processing of the Drosophila NF-ΚB Factor Relish. Proc. Natl. Acad. Sci. USA 2003, 100, 5991–5996. [Google Scholar] [CrossRef]
- Lemaitre, B.; Hoffmann, J. The Host Defense of Drosophila Melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef]
- Keebaugh, E.S.; Schlenke, T.A. Insights from Natural Host-Parasite Interactions: The Drosophila Model. Dev. Comp. Immunol. 2014, 42, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Tafesh-Edwards, G.; Eleftherianos, I. JNK Signaling in Drosophila Immunity and Homeostasis. Immunol. Lett. 2020, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.C.; Ley, S.C. Mitogen-Activated Protein Kinases in Innate Immunity. Nat. Rev. Immunol. 2013, 679–692. [Google Scholar] [CrossRef]
- Goberdhan, D.C.I.; Wilson, C. JNK, Cytoskeletal Regulator and Stress Response Kinase? A Drosophila Perspective. BioEssays. 1998, 1009–1019. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 1911–1912. [Google Scholar] [CrossRef]
- Stronach, B. Dissecting JNK Signaling, One KKKinase at a Time. Dev. Dyn. 2005, 575–584. [Google Scholar] [CrossRef]
- Biteau, B.; Karpac, J.; Hwangbo, D.S.; Jasper, H. Regulation of Drosophila Lifespan by JNK Signaling. Exp. Gerontol. 2011, 46, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Pinal, N.; Calleja, M.; Morata, G. Pro-Apoptotic and pro-Proliferation Functions of the JNK Pathway of Drosophila: Roles in Cell Competition, Tumorigenesis and Regeneration. Open Biol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Jajere, S.M. A Review of Salmonella Enterica with Particular Focus on the Pathogenicity and Virulence Factors, Host Specificity and Adaptation and Antimicrobial Resistance Including Multidrug Resistance. Vet. World 2019, 504–521. [Google Scholar] [CrossRef]
- Collier-Hyams, L.S.; Zeng, H.; Sun, J.; Tomlinson, A.D.; Bao, Z.Q.; Chen, H.; Madara, J.L.; Orth, K.; Neish, A.S. Cutting Edge: Salmonella AvrA Effector Inhibits the Key Proinflammatory, Anti-Apoptotic NF-ΚB Pathway. J. Immunol. 2002, 169, 2846–2850. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef]
- Wu, H.; Jones, R.M.; Neish, A.S. The Salmonella Effector AvrA Mediates Bacterial Intracellular Survival during Infection in Vivo. Cell. Microbiol. 2012, 14, 28–39. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Y.G.; Xia, Y.; Xu, X.; Jiao, X.; Sun, J. Salmonella Enteritidis Effector AvrA Stabilizes Intestinal Tight Junctions via the JNK Pathway. J. Biol. Chem. 2016, 291, 26837–26849. [Google Scholar] [CrossRef]
- Menanteau-Ledouble, S.; Kumar, G.; Saleh, M.; El-Matbouli, M. Aeromonas Salmonicida: Updates on an Old Acquaintance. Dis. Aquat. Org. 2016, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Varshney, A.; Das, M.; Chaudhary, P.; Kumari, R.; Yadav, K. Aeromonas Salmonicida as a Causative Agent for Postoperative Endophthalmitis. Middle East Afr. J. Ophthalmol. 2017, 24, 213–215. [Google Scholar] [CrossRef]
- Tewari, R.; Dudeja, M.; Nandy, S.; Das, A.K. Isolation of Aeromonas Salmonicida from Human Blood Sample: A Case Report. J. Clin. Diagn. Res. 2014, 8, 139–140. [Google Scholar] [CrossRef]
- Fehr, D.; Casanova, C.; Liverman, A.; Blazkova, H.; Orth, K.; Dobbelaere, D.; Frey, J.; Burr, S.E. AopP, a Type III Effector Protein of Aeromonas Salmonicida, Inhibits the NF-ΚB Signalling Pathway. Microbiology 2006, 152, 2809–2818. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Luo, L.; Moberg, K.H. Aeromonas Salmonicida-Secreted Protein AopP Is a Potent Inducer of Apoptosis in a Mammalian and a Drosophila Model. Cell. Microbiol. 2012, 14, 274–285. [Google Scholar] [CrossRef]
- Prod’homme, M.; Micol, L.A.; Weitsch, S.; Gassend, J.L.; Martinet, O.; Bellini, C. Cutaneous Infection and Bactaeremia Caused by Erwinia Billingiae: A Case Report. New Microbes New Infect. 2017, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Verdonck, K.; González, E.; Van Dooren, S.; Vandamme, A.M.; Vanham, G.; Gotuzzo, E. Human T-Lymphotropic Virus 1: Recent Knowledge about an Ancient Infection. Lancet Infect. Dis. 2007, 266–281. [Google Scholar] [CrossRef]
- Hermine, O.; Ramos, J.C.; Tobinai, K. A Review of New Findings in Adult T-Cell Leukemia–Lymphoma: A Focus on Current and Emerging Treatment Strategies. Adv. Ther. 2018, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Giam, C.Z. NF-ΚB Signaling Mechanisms in HTLV-1-Induced Adult T-Cell Leukemia/Lymphoma. FEBS J. 2018, 3324–3336. [Google Scholar] [CrossRef] [PubMed]
- Journo, C.; Bonnet, A.; Favre-Bonvin, A.; Turpin, J.; Vinera, J.; Cote, E.; Chevalier, S.A.; Kfoury, Y.; Bazarbachi, A.; Pique, C.; et al. Human T Cell Leukemia Virus Type 2 Tax-Mediated NF-κB Activation Involves a Mechanism Independent of Tax Conjugation to Ubiquitin and SUMO. J. Virol. 2013, 87, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Bex, F.; Murphy, K.; Wattiez, R.; Burny, A.; Gaynor, R.B. Phosphorylation of the Human T-Cell Leukemia Virus Type 1 Transactivator Tax on Adjacent Serine Residues Is Critical for Tax Activation. J. Virol. 1999, 73, 738–745. [Google Scholar] [CrossRef]
- Hleihel, R.; Khoshnood, B.; Dacklin, I.; Omran, H.; Mouawad, C.; Dassouki, Z.; El-Sabban, M.; Shirinian, M.; Grabbe, C. The HTLV-1 Oncoprotein Tax Is Modified by the Ubiquitin Related Modifier 1 (Urm1). Retrovirology 2018, 15. [Google Scholar] [CrossRef]
- Pedrioli, P.G.A.; Leidel, S.; Hofmann, K. Urm1 at the Crossroad of Modifications. “Protein Modifications: Beyond the Usual Suspects” Review Series. EMBO Rep. 2008, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Shirinian, M.; Kambris, Z.; Hamadeh, L.; Grabbe, C.; Journo, C.; Mahieux, R.; Bazarbachi, A. A Transgenic Drosophila Melanogaster Model To Study Human T-Lymphotropic Virus Oncoprotein Tax-1-Driven Transformation In Vivo. J. Virol. 2015, 89, 8092–8095. [Google Scholar] [CrossRef] [PubMed]
- Shirinian, M.; Kfoury, Y.; Dassouki, Z.; El-Hajj, H.; Bazarbachi, A. Tax-1 and Tax-2 Similarities and Differences: Focus on Post-Translational Modifications and NF-ΚB Activation. Front. Microbiol. 2013. [Google Scholar] [CrossRef]
- Higuchi, M.; Fujii, M. Distinct Functions of HTLV-1 Tax1 from HTLV-2 Tax2 Contribute Key Roles to Viral Pathogenesis. Retrovirology 2009. [Google Scholar] [CrossRef] [PubMed]
- Motai, Y.; Takahashi, M.; Takachi, T.; Higuchi, M.; Hara, T.; Mizuguchi, M.; Aoyagi, Y.; Terai, S.; Tanaka, Y.; Fujii, M. Human T-Cell Leukemia Virus Type 1 (HTLV-1) Tax1 Oncoprotein but Not HTLV-2 Tax2 Induces the Expression of OX40 Ligand by Interacting with P52/P100 and RelB. Virus Genes 2016, 52, 4–13. [Google Scholar] [CrossRef]
- Moir, S.; Fauci, A.S. B-Cell Responses to HIV Infection. Immunol. Rev. 2017, 33–48. [Google Scholar] [CrossRef]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and Secondary Structure of an Entire HIV-1 RNA Genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef]
- González, M.E. Vpu Protein: The Viroporin Encoded by HIV-1. Viruses 2015, 4352–4368. [Google Scholar] [CrossRef]
- Akari, H.; Bour, S.; Kao, S.; Adachi, A.; Strebel, K. The Human Immunodeficiency Virus Type 1 Accessory Protein Vpu Induces Apoptosis by Suppressing the Nuclear Factor ΚB-Dependent Expression of Antiapoptotic Factors. J. Exp. Med. 2001, 194, 1299–1311. [Google Scholar] [CrossRef]
- Bour, S.; Perrin, C.; Akari, H.; Strebel, K. The Human Immunodeficiency Virus Type 1 Vpu Protein Inhibits NF-ΚB Activation by Interfering with ΒTrCP-Mediated Degradation of IκB. J. Biol. Chem. 2001, 276, 15920–15928. [Google Scholar] [CrossRef] [PubMed]
- Leulier, F.; Marchal, C.; Miletich, I.; Limbourg-Bouchon, B.; Benarous, R.; Lemaitre, B. Directed Expression of the HIV-1 Accessory Protein Vpu in Drosophila Fat-Body Cells Inhibits Toll-Dependent Immune Responses. Embo Rep. 2003, 4, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Limper, A.H.; Adenis, A.; Le, T.; Harrison, T.S. Fungal Infections in HIV/AIDS. Lancet Infect. Dis. 2017, e334–e343. [Google Scholar] [CrossRef]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; Jesus, P.D.D.; Herbert, K.M.; Pache, L.; Smith, N.; Der Merwe, J.A.V.; et al. Hiv-1 Vpu Is a Potent Transcriptional Suppressor of Nf-Kb-Elicited Antiviral Immune Responses. Elife 2019, 8. [Google Scholar] [CrossRef]
- Chan, D.C.; Kim, P.S. HIV Entry and Its Inhibition. Cell 1998, 681–684. [Google Scholar] [CrossRef]
- Kirchhoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Absence of Intact Nef Sequences in a Long-Term Survivor with Nonprogressive HIV-1 Infection. N. Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef]
- Hanna, Z.; Kay, D.G.; Rebai, N.; Guimond, A.; Jothy, S.; Jolicoeur, P. Nef Harbors a Major Determinant of Pathogenicity for an AIDS-like Disease Induced by HIV-1 in Transgenic Mice. Cell 1998, 95, 163–175. [Google Scholar] [CrossRef]
- Udenwobele, D.I.; Su, R.C.; Good, S.V.; Ball, T.B.; Shrivastav, S.V.; Shrivastav, A. Myristoylation: An Important Protein Modification in the Immune Response. Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Lee, S.B.; Park, J.; Jung, J.U.; Chung, J. Nef Induces Apoptosis by Activating JNK Signaling Pathway and Inhibits NF-ΚB-Dependent Immune Responses in Drosophila. J. Cell Sci. 2005, 118, 1851–1859. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kounatidis, I.; Ligoxygakis, P. Drosophila as a Model to Study the Role of Blood Cells in Inflammation, Innate Immunity and Cancer. Front. Cell. Infect. Microbiol. 2014. [Google Scholar] [CrossRef]
- Barber, G.N. Host Defense, Viruses and Apoptosis. Cell Death Differ. 2001, 113–126. [Google Scholar] [CrossRef]
- Steller, H. Regulation of Apoptosis in Drosophila. Cell Death Differ. 2008, 1132–1138. [Google Scholar] [CrossRef]
- Berthelet, J.; Dubrez, L. Regulation of Apoptosis by Inhibitors of Apoptosis (IAPs). Cells 2013, 2, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017. [Google Scholar] [CrossRef]
- Goehring, U.M.; Schmidt, G.; Pederson, K.J.; Aktories, K.; Barbieri, J.T. The N-Terminal Domain of Pseudomonas aeruginosa Exoenzyme S Is a GTPase- Activating Protein for Rho GTPases. J. Biol. Chem. 1999, 274, 36369–36372. [Google Scholar] [CrossRef]
- Huber, P.; Bouillot, S.; Elsen, S.; Attrée, I. Sequential Inactivation of Rho GTPases and Lim Kinase by Pseudomonas aeruginosa Toxins ExoS and ExoT Leads to Endothelial Monolayer Breakdown. Cell. Mol. Life Sci. 2014, 71, 1927–1941. [Google Scholar] [CrossRef]
- Hauser, A.R. Ventilator-Associated Pneumonia Caused by Pseudomonas aeruginosa: Cap Your Needles! Crit. Care Med. 2012, 2503–2504. [Google Scholar] [CrossRef]
- Avet-rochex, A.; Perrin, J.; Bergeret, E.; Fauvarque, M.O. Rac2 Is a Major Actor of Drosophila Resistance to Pseudomonas aeruginosa Acting in Phagocytic Cells. Genes Cells 2007, 12, 1193–1204. [Google Scholar] [CrossRef]
- Rangel, S.M.; Diaz, M.H.; Knoten, C.A.; Zhang, A.; Hauser, A.R. The Role of ExoS in Dissemination of Pseudomonas aeruginosa during Pneumonia. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.C.C.; Lau, S.K.P.; Woo, P.C.Y.; Kwok, Y.Y. Severe Acute Respiratory Syndrome Coronavirus as an Agent of Emerging and Reemerging Infection. Clin. Microbiol. Rev. 2007, 660–694. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.; et al. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Marra, M.A.; Jones, S.J.M.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.N.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome Sequence of the SARS-Associated Coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a Possible Cause of Severe Acute Respiratory Syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Nicholls, J.; Dong, X.P.; Jiang, G.; Peiris, M. SARS: Clinical Virology and Pathogenesis. Respirology 2003. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Murakami, M.; Hirano, T.; Saijo, M.; Kurane, I.; Morikawa, S. Tyrosine Dephosphorylation of STAT3 in SARS Coronavirus-Infected Vero E6 Cells. FEBS Lett. 2004, 577, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. JNK and PI3k/Akt Signaling Pathways Are Required for Establishing Persistent SARS-CoV Infection in Vero E6 Cells. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1741, 4–10. [Google Scholar] [CrossRef]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of P38 MAPK and Its Downstream Targets in SARS Coronavirus-Infected Cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef]
- Hu, Y.; Wen, J.; Tang, L.; Zhang, H.; Zhang, X.; Li, Y.; Wang, J.; Han, Y.; Li, G.; Shi, J.; et al. The M Protein of SARS-CoV: Basic Structural and Immunological Properties. Genom. Proteom. Bioinforma. Beijing Genom. Inst. 2003, 1, 118–130. [Google Scholar] [CrossRef]
- Yu, C.J.; Chen, Y.C.; Hsiao, C.H.; Kuo, T.C.; Chang, S.C.; Lu, C.Y.; Wei, W.C.; Lee, C.H.; Huang, L.M.; Chang, M.F.; et al. Identification of a Novel Protein 3a from Severe Acute Respiratory Syndrome Coronavirus. FEBS Lett. 2004, 565, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Ma, C.W.; Chan, W.Y.; Chan, H.Y.E. The SARS-Coronavirus Membrane Protein Induces Apoptosis through Modulating the Akt Survival Pathway. Arch. Biochem. Biophys. 2007, 459, 197–207. [Google Scholar] [CrossRef]
- Tsoi, H.; Li, L.; Chen, Z.S.; Lau, K.F.; Tsui, S.K.W.; Chan, H.Y.E. The SARS-Coronavirus Membrane Protein Induces Apoptosis via Interfering with PDK1PKB/Akt Signalling. Biochem. J. 2014, 464, 439–447. [Google Scholar] [CrossRef]
- Wong, S.L.A.; Chen, Y.; Chak, M.C.; Chan, C.S.M.; Chan, P.K.S.; Chui, Y.L.; Kwok, P.F.; Waye, M.M.Y.; Tsui, S.K.W.; Chan, H.Y.E. In Vivo Functional Characterization of the SARS-Coronavirus 3a Protein in Drosophila. Biochem. Biophys. Res. Commun. 2005, 337, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Tsoi, H.; Chan, W.M.; Zhai, S.; Wong, C.O.; Yao, X.; Chan, W.Y.; Tsui, S.K.W.; Chan, H.Y.E. The Ion Channel Activity of the SARS-Coronavirus 3a Protein Is Linked to Its pro-Apoptotic Function. Int. J. Biochem. Cell Biol. 2009, 41, 2232–2239. [Google Scholar] [CrossRef]
- Yang, S.; Tian, M.; Johnson, A.N. SARS-CoV-2 Protein ORF3a Is Pathogenic in Drosophila and Causes Phenotypes Associated with COVID-19 Post-Viral Syndrome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Shabbir, S.; Udin, N.; Aziz, B.; Amanullah, A.; Raza, F.; Baig, A.A. Dataset for Homologous Proteins in Drosophila Melanogaster for SARS-CoV-2/Human Interactome. Data Br. 2020, 32. [Google Scholar] [CrossRef]
- Marchal, C.; Vinatier, G.; Sanial, M.; Plessis, A.; Pret, A.M.; Limbourg-Bouchon, B.; Théodore, L.; Netter, S. The HIV-1 Vpu Protein Induces Apoptosis in Drosophila via Activation of JNK Signaling. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Hashim, M.J. Global Burden of Deaths from Epstein-Barr Virus Attributable Malignancies 1990-2010. Infect. Agent. Cancer 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr Virus Lytic Program Is Controlled by the Co-Operative Functions of Two Transactivators. Embo J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J.; Rowe, D.T.; Rooney, C.M.; Kouzarides, T. Epstein-Barr Virus BZLF1 Trans-Activator Specifically Binds to a Consensus AP-1 Site and Is Related to c-Fos. Embo J. 1989, 8, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.L.; Kenney, S. The Epstein-Barr Virus BZLF1 Protein Interacts Physically and Functionally with the Histone Acetylase CREB-Binding Protein. J. Virol. 1999, 73, 6551–6558. [Google Scholar] [CrossRef]
- Adamson, A.L.; Wright, N.; LaJeunesse, D.R. Modeling Early Epstein-Barr Virus Infection in Drosophila Melanogaster: The BZLF1 Protein. Genetics 2005, 171, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Mauser, A.; Holley-Guthrie, E.; Zanation, A.; Yarborough, W.; Kaufmann, W.; Klingelhutz, A.; Seaman, W.T.; Kenney, S. The Epstein-Barr Virus Immediate-Early Protein BZLF1 Induces Expression of E2F-1 and Other Proteins Involved in Cell Cycle Progression in Primary Keratinocytes and Gastric Carcinoma Cells. J. Virol. 2002, 76, 12543–12552. [Google Scholar] [CrossRef]
- Kavaler, J.; Fu, W.; Duan, H.; Noll, M.; Posakony, J.W. An Essential Role for the Drosophila Pax2 Homolog in the Differentiation of Adult Sensory Organs. Development 1999, 126, 2261–2272. [Google Scholar] [PubMed]
- Chen, Y.L.; Chen, Y.J.; Tsai, W.H.; Ko, Y.C.; Chen, J.Y.; Lin, S.F. The Epstein-Barr Virus Replication and Transcription Activator, Rta/BRLF1, Induces Cellular Senescence in Epithelial Cells. Cell Cycle 2009, 8, 58–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swenson, J.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr Virus Immediate-Early Protein BRLF1 Interacts with CBP, Promoting Enhanced BRLF1 Transactivation. J. Virol. 2001, 75, 6228–6234. [Google Scholar] [CrossRef] [PubMed]
- Zacny, V.L.; Wilson, J.; Pagano, J.S. The Epstein-Barr Virus Immediate-Early Gene Product, BRLF1, Interacts with the Retinoblastoma Protein during the Viral Lytic Cycle. J. Virol. 1998, 72, 8043–8051. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.I.H. Zika Virus Infection in Man. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 335–338. [Google Scholar] [CrossRef]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastãre, S.; Valour, F.; Baudouin, L.; Mallet, H.P.; Musso, D.; Ghawche, F. Zika Virus Infection Complicated by Guillain-Barré Syndrome Acase Report, French Polynesia, December 2013. Eurosurveillance 2014, 19. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Malkki, H. CNS Infections: Mouse Studies Confirm the Link between Zika Virus Infection and Microcephaly. Nat. Rev. Neurol. 2016, 369. [Google Scholar] [CrossRef]
- Peçanha, P.M.; Gomes Junior, S.C.; Pone, S.M.; da Pone, M.V.S.; Vasconcelos, Z.; Zin, A.; Vilibor, R.H.H.; Costa, R.P.; Meio, M.D.B.B.; Nielsen-Saines, K.; et al. Neurodevelopment of Children Exposed Intra-Uterus by Zika Virus: A Case Series. PLoS ONE 2020, 15, e0229434. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Jaiswal, M.; Charng, W.L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A Drosophila Genetic Resource of Mutants to Study Mechanisms Underlying Human Genetic Diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Maddirevula, S.; Ewida, N.; Alsahli, S.; Abdel-Salam, G.M.H.; Zaki, M.S.; Al Tala, S.; Alhashem, A.; Softah, A.; Al-Owain, M.; et al. Genomic and Phenotypic Delineation of Congenital Microcephaly. Genet. Med. 2019, 21, 545–552. [Google Scholar] [CrossRef]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell 2019, 51, 713–729.e6. [Google Scholar] [CrossRef]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Ankle2, a Target of Zika Virus, Controls Asymmetric Cell Division of Neuroblasts and Uncovers a Novel Microcephaly Pathway. Ssrn Electron. J. 2019. [Google Scholar] [CrossRef]
- Renbaum, P.; Kellerman, E.; Jaron, R.; Geiger, D.; Segel, R.; Lee, M.; King, M.C.; Levy-Lahad, E. Spinal Muscular Atrophy with Pontocerebellar Hypoplasia Is Caused by a Mutation in the VRK1 Gene. Am. J. Hum. Genet. 2009, 85, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga-Jauregui, C.; Lotze, T.; Jamal, L.; Penney, S.; Campbell, I.M.; Pehlivan, D.; Hunter, J.V.; Woodbury, S.L.; Raymond, G.; Adesina, A.M.; et al. Mutations in VRK1 Associated with Complex Motor and Sensory Axonal Neuropathy plus Microcephaly. JAMA Neurol. 2013, 70, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.M.; McGavran, L.; Robinson, J.; Waldstein, G.; Macfarlane, J.; Zonona, J.; Reiss, J.; Lahr, M.; Allen, L.; Magenis, E. Interstitial Deletion of (17)(P11.2p11.2) in Nine Patients. Am. J. Med. Genet. 1986, 24, 393–414. [Google Scholar] [CrossRef]
- Rossi, S.L.; Ebel, G.D.; Shan, C.; Shi, P.Y.; Vasilakis, N. Did Zika Virus Mutate to Cause Severe Outbreaks? Trends Microbiol. 2018, 877–885. [Google Scholar] [CrossRef]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68.e3. [Google Scholar] [CrossRef]
- Harsh, S.; Ozakman, Y.; Kitchen, S.M.; Paquin-Proulx, D.; Nixon, D.F.; Eleftherianos, I. Dicer-2 Regulates Resistance and Maintains Homeostasis against Zika Virus Infection in Drosophila. J. Immunol. 2018, 201, 3058–3072. [Google Scholar] [CrossRef]
- Harsh, S.; Fu, Y.; Kenney, E.; Han, Z.; Eleftherianos, I. Zika Virus Non-Structural Protein NS4A Restricts Eye Growth in Drosophila through Regulation of JAK/STAT Signaling. Dmm Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat. Immunol. 2017, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Agaisse, H.; Perrimon, N. The Roles of JAK/STAT Signaling in Drosophila Immune Responses. Immunol. Rev. 2004, 72–82. [Google Scholar] [CrossRef]
- Salazar, J.L.; Yamamoto, S. Integration of Drosophila and Human Genetics to Understand Notch Signaling Related Diseases. Adv. Exp. Med. Biol. 2018, 1066, 141–185. [Google Scholar] [CrossRef]
- Radtke, F.; Fasnacht, N.; MacDonald, H.R. Notch Signaling in the Immune System. Immunity 2010, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.L.; Yang, S.A.; Yamamoto, S. Post-Developmental Roles of Notch Signaling in the Nervous System. Biomolecules 2020, 10, 985. [Google Scholar] [CrossRef]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A Single Mutation in the PrM Protein of Zika Virus Contributes to Fetal Microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An Evolutionary NS1 Mutation Enhances Zika Virus Evasion of Host Interferon Induction. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 647–671. [Google Scholar] [CrossRef]
- Inglesby, T.V.; O’Toole, T.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Friedlander, A.M.; Gerberding, J.; Hauer, J.; Hughes, J.; et al. Anthrax as a Biological Weapon, 2002: Updated Recommendations for Management. J. Am. Med. Assoc. 2002, 2236–2252. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 185–208. [Google Scholar] [CrossRef]
- Pezard, C.; Berche, P.; Mock, M. Contribution of Individual Toxin Components to Virulence of Bacillus Anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar] [CrossRef]
- Guichard, A. Common Target Anthrax Lethal Factor & Edema Factor. Proc. Natl. Acad. Sci. USA 2006, 103, 3244. [Google Scholar] [PubMed]
- Agnès, F.; Suzanne, M.; Noselli, S. The Drosophila JNK Pathway Controls the Morphogenesis of Imaginal Discs during Metamorphosis. Development 1999, 126, 5453–5462. [Google Scholar]
- Guichard, A.; Park, J.M.; Cruz-Moreno, B.; Karin, M.; Bier, E. Anthrax Lethal Factor and Edema Factor Act on Conserved Targets in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 3244–3249. [Google Scholar] [CrossRef] [PubMed]
- Blair, S.S. Wing Vein Patterning in Drosophila and the Analysis of Intercellular Signaling. Annu. Rev. Cell Dev. Biol. 2007, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Okabe, M.; Tanimura, T.; Mikoshiba, K.; Nishida, Y.; Okano, H. The Drosophila Secreted Protein Argos Regulates Signal Transduction in the Ras/MAPK Pathway. Dev. Biol. 1996, 178, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.P.; Boone, S.A.; Liang, X.; Duesbery, N.S. Anthrax Lethal Factor Proteolysis and Inactivation of MAPK Kinase. J. Biol. Chem. 2003, 278, 9402–9406. [Google Scholar] [CrossRef] [PubMed]
- Hartl, T.A.; Scott, M.P. Wing Tips: The Wing Disc as a Platform for Studying Hedgehog Signaling. Methods 2014, 68, 199–206. [Google Scholar] [CrossRef]
- Leppla, S.H. Anthrax Toxin Edema Factor: A Bacterial Adenylate Cyclase That Increases Cyclic AMP Concentrations in Eukaryotic Cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef]
- Lum, L.; Beachy, P.A. The Hedgehog Response Network: Sensors, Switches, and Routers. Science 2004, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Kiger, J.A.; Eklund, J.L.; Younger, S.H.; O’Kane, C.J. Transgenic Inhibitors Identify Two Roles for Protein Kinase A in Drosophila Development. Genetics 1999, 152, 281–290. [Google Scholar] [PubMed]
- Guichard, A.; McGillivray, S.M.; Cruz-Moreno, B.; Van Sorge, N.M.; Nizet, V.; Bier, E. Anthrax Toxins Cooperatively Inhibit Endocytic Recycling by the Rab11/Sec15 Exocyst. Nature 2010, 467, 854–858. [Google Scholar] [CrossRef]
- Giagtzoglou, N.; Yamamoto, S.; Zitserman, D.; Graves, H.K.; Schulze, K.L.; Wang, H.; Klein, H.; Roegiers, F.; Bellen, H.J. DEHBP1 Controls Exocytosis and Recycling of Delta during Asymmetric Divisions. J. Cell Biol. 2012, 196, 65–83. [Google Scholar] [CrossRef]
- Jafar-Nejad, H.; Andrews, H.K.; Acar, M.; Bayat, V.; Wirtz-Peitz, F.; Mehta, S.Q.; Knoblich, J.A.; Bellen, H.J. Sec15, a Component of the Exocyst, Promotes Notch Signaling during the Asymmetric Division of Drosophila Sensory Organ Precursors. Dev. Cell 2005, 9, 351–363. [Google Scholar] [CrossRef]
- Guichard, A.; Jain, P.; Moayeri, M.; Schwartz, R.; Chin, S.; Zhu, L.; Cruz-Moreno, B.; Liu, J.Z.; Aguilar, B.; Hollands, A.; et al. Anthrax Edema Toxin Disrupts Distinct Steps in Rab11-Dependent Junctional Transport. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac Is a Rap1 Guanine-Nucleotide-Exchange Factor Directly Activated by Cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Balzac, F.; Avolio, M.; Degani, S.; Kaverina, I.; Torti, M.; Silengo, L.; Small, J.V.; Retta, S.F. E-Cadherin Endocytosis Regulates the Activity of Rap1: A Traffic Light GTPase at the Crossroads between Cadherin and Integrin Function. J. Cell Sci. 2005, 118, 4765–4783. [Google Scholar] [CrossRef]
- Weil, A.A.; Harris, J.B. Vibrio Cholerae. In Molecular Medical Microbiology, 2nd ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 2–3, pp. 1079–1098. [Google Scholar] [CrossRef]
- Dick, M.H.; Guillerm, M.; Moussy, F.; Chaignat, C.L. Review of Two Decades of Cholera Diagnostics—How Far Have We Really Come? PLoS Negl. Trop. Dis. 2012. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R Domain by CAMP-Dependent Protein Kinase Regulates the CFTR Chloride Channel. Cell 1991, 66, 1027–1036. [Google Scholar] [CrossRef]
- Guichard, A.; Cruz-Moreno, B.C.; Aguilar, B.; Van Sorge, N.M.; Kuang, J.; Kurkciyan, A.A.; Wang, Z.; Hang, S.; Pineton De Chambrun, G.P.; McCole, D.F.; et al. Cholera Toxin Disrupts Barrier Function by Inhibiting Exocyst-Mediated Trafficking of Host Proteins to Intestinal Cell Junctions. Cell Host Microbe 2013, 14, 294–305. [Google Scholar] [CrossRef]
- Holmgren, J.; Lonnroth, I.; Svennerholm, L. Tissue Receptor for Cholera Exotoxin: Postulated Structure from Studies with G(M1) Ganglioside and Related Glycolipids. Infect. Immun. 1973, 8, 208–214. [Google Scholar] [CrossRef] [PubMed]
- De Haan, L.; Hirst, T.R. Cholera Toxin: A Paradigm for Multi-Functional Engagement of Cellular Mechanisms (Review). Mol. Membr. Biol. 2004, 77–92. [Google Scholar] [CrossRef]
- Rera, M.; Bahadorani, S.; Cho, J.; Koehler, C.L.; Ulgherait, M.; Hur, J.H.; Ansari, W.S.; Lo, T.; Jones, D.L.; Walker, D.W. Modulation of Longevity and Tissue Homeostasis by the Drosophila PGC-1 Homolog. Cell Metab. 2011, 14, 623–634. [Google Scholar] [CrossRef]
- Blow, N.S.; Salomon, R.N.; Garrity, K.; Reveillaud, I.; Kopin, A.; Jackson, F.R.; Watnick, P.I. Vibrio Cholerae Infection of Drosophila Melanogaster Mimics the Human Disease Cholera. PLoS Pathog. 2005, 1, e8. [Google Scholar] [CrossRef] [PubMed]
- Crough, T.; Khanna, R. Immimobiology of Human Cytomegalovirus: From Bench to Bedside. Clin. Microbiol. Rev. 2009, 76–98. [Google Scholar] [CrossRef]
- Landolfo, S.; Gariglio, M.; Gribaudo, G.; Lembo, D. The Human Cytomegalovirus. Pharmacol. Ther. 2003, 269–297. [Google Scholar] [CrossRef]
- Jaskoll, T.; Abichaker, G.; Sedghizadeh, P.P.; Bringas, P.; Melnick, M. Cytomegalovirus Induces Abnormal Chondrogenesis and Osteogenesis during Embryonic Mandibular Development. BMC Dev. Biol. 2008, 8. [Google Scholar] [CrossRef]
- Schleiss, M.R. Nonprimate Models of Congenital Cytomegalovirus (CMV) Infection: Gaining Insight into Pathogenesis and Prevention of Disease in Newborns. ILAR J. 2006, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F.; Meier, J.L. Immediate–Early Viral Gene Regulation and Function; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Isomura, H.; Stinski, M.F. The Human Cytomegalovirus Major Immediate-Early Enhancer Determines the Efficiency of Immediate-Early Gene Transcription and Viral Replication in Permissive Cells at Low Multiplicity of Infection. J. Virol. 2003, 77, 3602–3614. [Google Scholar] [CrossRef]
- Isomura, H.; Tsurumi, T.; Stinski, M.F. Role of the Proximal Enhancer of the Major Immediate-Early Promoter in Human Cytomegalovirus Replication. J. Virol. 2004, 78, 12788–12799. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Steinberg, R.; Shemer-Avni, Y.; Adler, N.; Neuman-Silberberg, S. Human Cytomegalovirus Immediate-Early-Gene Expression Disrupts Embryogenesis in Transgenic Drosophila. Transgenic Res. 2008, 17, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Tepass, U.; Tanentzapf, G. Epithelial Cell Polarity and Cell Junctions in Drosophila. Annu. Rev. Genet. 2001, 35, 747–784. [Google Scholar] [CrossRef]
- Chen, D.H.; Jiang, H.; Lee, M.; Liu, F.; Zhou, Z.H. Three-Dimensional Visualization of Tegument/Capsid Interactions in the Intact Human Cytomegalovirus. Virology 1999, 260, 10–16. [Google Scholar] [CrossRef]
- Bentz, G.L.; Jarquin-Pardo, M.; Chan, G.; Smith, M.S.; Sinzger, C.; Yurochko, A.D. Human Cytomegalovirus (HCMV) Infection of Endothelial Cells Promotes Naïve Monocyte Extravasation and Transfer of Productive Virus To Enhance Hematogenous Dissemination of HCMV. J. Virol. 2006, 80, 11539–11555. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Blaser, M.J. The Role of CagA in the Gastric Biology of Helicobacter Pylori. Cancer Res. 2016, 4028–4031. [Google Scholar] [CrossRef]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.-I.; Kook, M.-C.; Park, B.; Joo, J. Family History of Gastric Cancer and Helicobacter Pylori Treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Higashi, H. Helicobacter Pylori CagA: A New Paradigm for Bacterial Carcinogenesis. Cancer Sci. 2005, 835–843. [Google Scholar] [CrossRef]
- Suzuki, M.; Mimuro, H.; Suzuki, T.; Park, M.; Yamamoto, T.; Sasakawa, C. Interaction of CagA with Crk Plays an Important Role in Helicobacter Pylori-Induced Loss of Gastric Epithelial Cell Adhesion. J. Exp. Med. 2005, 202, 1235–1247. [Google Scholar] [CrossRef]
- Churin, Y.; Al-Ghoul, L.; Kepp, O.; Meyer, T.F.; Birchmeier, W.; Naumann, M. Helicobacter Pylori CagA Protein Targets the C-Met Receptor and Enhances the Motogenic Response. J. Cell Biol. 2003, 161, 249–255. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and Function of Helicobacter Pylori Caga, the First-Identified Bacterial Protein Involved in Human Cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 196–219. [Google Scholar] [CrossRef]
- Mavromatakis, Y.E.; Tomlinson, A. Switching Cell Fates in the Developing Drosophila Eye. Development 2013, 140, 4353–4361. [Google Scholar] [CrossRef]
- Lusk, J.B.; Lam, V.Y.M.; Tolwinski, N.S. Epidermal Growth Factor Pathway Signaling in Drosophila Embryogenesis: Tools for Understanding Cancer. Cancers 2017, 9, 16. [Google Scholar] [CrossRef]
- Tomlinson, A.; Mavromatakis, Y.E.; Arias, R. The Role of Sevenless in Drosophila R7 Photoreceptor Specification. Dev. Biol. 2019, 454, 181–189. [Google Scholar] [CrossRef]
- Raabe, T. The Sevenless Signaling Pathway: Variations of a Common Theme. Biochim. Biophys. Acta Mol. Cell Res. 2000, 151–163. [Google Scholar] [CrossRef]
- Baker, N.E.; Rubin, G.M. Effect on Eye Development of Dominant Mutations in Drosophila Homologue of the EGF Receptor. Nature 1989, 340, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.A.; Bowtell, D.D.L.; Dodson, G.S.; Laverty, T.R.; Rubin, G.M. Ras1 and a Putative Guanine Nucleotide Exchange Factor Perform Crucial Steps in Signaling by the Sevenless Protein Tyrosine Kinase. Cell 1991, 67, 701–716. [Google Scholar] [CrossRef]
- Botham, C.M.; Wandler, A.M.; Guillemin, K. A Transgenic Drosophila Model Demonstrates That the Helicobacter Pylori CagA Protein Functions as a Eukaryotic Gab Adaptor. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Rubin, G.M. Analysis of Genetic Mosaics in Developing and Adult Drosophila Tissues. Development 1993, 117, 1223–1237. [Google Scholar]
- Muyskens, J.B.; Guillemin, K. Helicobacter Pylori CagA Disrupts Epithelial Patterning by Activating Myosin Light Chain. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Selbach, M.; Moese, S.; Hurwitz, R.; Hauck, C.R.; Meyer, T.F.; Backert, S. The Helicobacter Pylori CagA Protein Induces Cortactin Dephosphorylation and Actin Rearrangement by C-Src Inactivation. Embo J. 2003, 22, 515–528. [Google Scholar] [CrossRef]
- Bourzac, K.M.; Botham, C.M.; Guillemin, K. Helicobacter Pylori CagA Induces AGS Cell Elongation through a Cell Retraction Defect That Is Independent of Cdc42, Rac1, and Arp2/3. Infect. Immun. 2007, 75, 1203–1213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, B.; Ceponis, P.J.M.; Sherman, P.M. Cytoskeletal Rearrangements in Gastric Epithelial Cells in Response to Helicobacter Pylori Infection. J. Med. Microbiol. 2003, 52, 861–867. [Google Scholar] [CrossRef][Green Version]
- Escudero, L.M.; Bischoff, M.; Freeman, M. Myosin II Regulates Complex Cellular Arrangement and Epithelial Architecture in Drosophila. Dev. Cell 2007, 13, 717–729. [Google Scholar] [CrossRef]
- Bresnick, A.R. Molecular Mechanisms of Nonmuscle Myosin-II Regulation. Curr. Opin. Cell Biol. 1999, 11, 26–33. [Google Scholar] [CrossRef]
- Wong, K.; Van Keymeulen, A.; Bourne, H.R. PDZRhoGEF and Myosin II Localize RhoA Activity to the Back of Polarizing Neutrophil-like Cells. J. Cell Biol. 2007, 179, 1141–1148. [Google Scholar] [CrossRef]
- Wandler, A.M.; Guillemin, K. Transgenic Expression of the Helicobacter Pylori Virulence Factor CagA Promotes Apoptosis or Tumorigenesis through JNK Activation in Drosophila. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Tateno, M.; Nishida, Y.; Adachi-Yamada, T. Regulation of JNK by Src during Drosophila Development. Science 2000, 287, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Igaki, T.; Miura, M. The Drosophila TNF Ortholog Eiger: Emerging Physiological Roles and Evolution of the TNF System. Semin. Immunol. 2014, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of Cell Polarity Drives Tumor Growth and Invasion through JNK Activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between RasV12 and Scribbled Clones Induces Tumour Growth and Invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef]
- Reid, D.W.; Muyskens, J.B.; Neal, J.T.; Gaddini, G.W.; Cho, L.Y.; Wandler, A.M.; Botham, C.M.; Guillemin, K. Identification of Genetic Modifiers of CagA-Induced Epithelial Disruption in Drosophila. Front. Cell. Infect. Microbiol. 2012, 2, 24. [Google Scholar] [CrossRef]
- Cani, P.D. Human Gut Microbiome: Hopes, Threats and Promises. Gut 2018, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.A.; Hernandez, D.Z.; Wong, Z.C.; Wandler, A.M.; Guillemin, K. The Bacterial Virulence Factor CagA Induces Microbial Dysbiosis That Contributes to Excessive Epithelial Cell Proliferation in the Drosophila Gut. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Nagase, L.; Hayashi, T.; Senda, T.; Hatakeyama, M. Dramatic Increase in SHP2 Binding Activity of Helicobacter Pylori Western CagA by EPIYA-C Duplication: Its Implications in Gastric Carcinogenesis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Palefsky, J.M. Human Papillomavirus Infections: Epidemiology and Disease Associations—Up To Date; UpToDate: Waltham, MA, USA, 2019; pp. 1–9. [Google Scholar]
- Handler, M.Z.; Handler, N.S.; Majewski, S.; Schwartz, R.A. Human Papillomavirus Vaccine Trials and Tribulations Clinical Perspectives. J. Am. Acad. Dermatol. 2015, 743–756. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of Incidence and Mortality of Cervical Cancer in 2018: A Worldwide Analysis. Lancet Glob. Heal. 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Narisawa-Saito, M.; Kiyono, T. Basic Mechanisms of High-Risk Human Papillomavirus-Induced Carcinogenesis: Roles of E6 and E7 Proteins. Cancer Sci. 2007, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.-M.; Galloway, D.A. Destabilization of the Retinoblastoma Tumor Suppressor by Human Papillomavirus Type 16 E7 Is Not Sufficient To Overcome Cell Cycle Arrest in Human Keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Wu, E.W.; Clemens, K.E.; Heck, D.V.; Münger, K. The Human Papillomavirus E7 Oncoprotein and the Cellular Transcription Factor E2F Bind to Separate Sites on the Retinoblastoma Tumor Suppressor Protein. J. Virol. 1993, 67, 2402–2407. [Google Scholar] [CrossRef]
- Pim, D.; Thomas, M.; Javier, R.; Gardiol, D.; Banks, L. HPV E6 Targeted Degradation of the Discs Large Protein: Evidence for the Involvement of a Novel Ubiquitin Ligase. Oncogene 2000, 19, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Kühne, C.; Gardiol, D.; Guarnaccia, C.; Amenitsch, H.; Banks, L. Differential Regulation of Human Papillomavirus E6 by Protein Kinase A: Conditional Degradation of Human Discs Large Protein by Oncogenic E6. Oncogene 2000, 19, 5884–5891. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Banks, L. A Systematic Analysis of Human Papillomavirus (HPV) E6 PDZ Substrates Identifies MAGI-1 as a Major Target of HPV Type 16 (HPV-16) and HPV-18 Whose Loss Accompanies Disruption of Tight Junctions. J. Virol. 2011, 85, 1757–1764. [Google Scholar] [CrossRef]
- Massimi, P.; Gammoh, N.; Thomas, M.; Banks, L. HPV E6 Specifically Targets Different Cellular Pools of Its PDZ Domain-Containing Tumour Suppressor Substrates for Proteasome-Mediated Degradation. Oncogene 2004, 23, 8033–8039. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP Regions That Direct Human Papillomavirus E6 Binding, Association with P53, and Ubiquitination of Associated Proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar] [CrossRef]
- Padash Barmchi, M.; Gilbert, M.; Thomas, M.; Banks, L.; Zhang, B.; Auld, V.J. A Drosophila Model of HPV E6-Induced Malignancy Reveals Essential Roles for Magi and the Insulin Receptor. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Kaku, M.; Simpson, D.M. HIV Neuropathy. Curr. Opin. HIV AIDS 2014, 521–526. [Google Scholar] [CrossRef]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat Protein Has a Versatile Role in Activating Viral Transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, M.; Zhou, S.; Zhou, Q. HIV-1 Tat Targets Microtubules to Induce Apoptosis, a Process Promoted by the pro-Apoptotic Bcl-2 Relative Bim. Embo J. 2002, 21, 6801–6810. [Google Scholar] [CrossRef]
- Romani, B.; Engelbrecht, S.; Glashoff, R.H. Functions of Tat: The Versatile Protein of Human Immunodeficiency Virus Type 1. J. Gen. Virol. 2010, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23. [Google Scholar] [CrossRef]
- Battaglia, P.A.; Zito, S.; Macchini, A.; Gigliani, F. A Drosophila Model of HIV-Tat-Related Pathogenicity. J. Cell Sci. 2001, 114, 2787–2794. [Google Scholar]
- Robinson, D.N.; Cooley, L. Genetic Analysis of the Actin Cytoskeleton in the Drosophila Ovary. Annu. Rev. Cell Dev. Biol. 1997, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, J.; Kalderon, D. Microtubule Polarity and Axis Formation in the Drosophila Oocyte. Dev. Dyn. 2006, 235, 1455–1468. [Google Scholar] [CrossRef]
- Wu, X.; Tanwar, P.S.; Raftery, L.A. Drosophila Follicle Cells: Morphogenesis in an Eggshell. Semin. Cell Dev. Biol. 2008, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, N.J.; Stephenson, E.C. Microtubules Mediate the Localization of Bicoid RNA during Drosophila Oogenesis. Development 1991, 113, 55–66. [Google Scholar] [PubMed]
- Weil, T.T.; Forrest, K.M.; Gavis, E.R. Localization of Bicoid MRNA in Late Oocytes Is Maintained by Continual Active Transport. Dev. Cell 2006, 11, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, M.E. Cytoplasmic Streaming in the Drosophila Oocyte. Annu. Rev. Cell Dev. Biol. 2016, 173–195. [Google Scholar] [CrossRef]
- Battaglia, P.A.; Ponti, D.; Naim, V.; Venanzi, S.; Psaila, R.; Gigliani, F. The HIV-Tat Protein Induces Chromosome Number Aberrations by Affecting Mitosis. Cell Motil. Cytoskelet. 2005, 61, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Jarboui, M.A.; Bidoia, C.; Woods, E.; Roe, B.; Wynne, K.; Elia, G.; Hall, W.W.; Gautier, V.W. Nucleolar Protein Trafficking in Response to HIV-1 Tat: Rewiring the Nucleolus. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Marasco, W.A.; Szilvay, A.M.; Kalland, K.H.; Helland, D.G.; Reyes, H.M.; Walter, R.J. Spatial Association of HIV-1 Tat Protein and the Nucleolar Transport Protein B23 in Stably Transfected Jurkat T-Cells. Arch. Virol. 1994, 139, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Troiano, M.; Bellenchi, G.; Battaglia, P.A.; Gigliani, F. The HIV Tat Protein Affects Processing of Ribosomal RNA Precursor. BMC Cell Biol. 2008, 9. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-Associated Neurocognitive Disorder—Pathogenesis and Prospects for Treatment. Nat. Rev. Neurol. 2016, 234–248. [Google Scholar] [CrossRef]
- Muñoz-Lasso, D.C.; Romá-Mateo, C.; Pallardó, F.V.; Gonzalez-Cabo, P. Much More Than a Scaffold: Cytoskeletal Proteins in Neurological Disorders. Cells 2020, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational Control in Synaptic Plasticity and Cognitive Dysfunction. Annu. Rev. Neurosci. 2014, 17–38. [Google Scholar] [CrossRef]
- Butel, J.S.; Lednicky, J.A. Cell and Molecular Biology of Simian Virus 40: Implications for Human Infections and Disease. J. Natl. Cancer Inst. 1999, 119–134. [Google Scholar] [CrossRef]
- Fisher, S.G.; Weber, L.; Carbone, M. Cancer Risk Associated with Simian Virus 40 Contaminated Polio Vaccine. Anticancer Res. 1999, 19, 2173–2180. [Google Scholar]
- Diamandopoulos, G.T. Leukemia, Lymphoma, and Osteosarcoma Induced in the Syrian Golden Hamster by Simian Virus 40. Science 1972, 176, 173–175. [Google Scholar] [CrossRef]
- Poulin, D.L.; DeCaprio, J.A. Is There a Role for SV40 in Human Cancer? J. Clin. Oncol. 2006, 4356–4365. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Mazzoni, E.; Bononi, I.; Tognon, M.; Martini, F. Association Between Simian Virus 40 and Human Tumors. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Garcea, R.L.; Imperiale, M.J. Simian Virus 40 Infection of Humans. J. Virol. 2003, 77, 5039–5045. [Google Scholar] [CrossRef] [PubMed]
- Skoczylas, C.; Henglein, B.; Rundell, K. PP2A-Dependent Transactivation of the Cyclin A Promoter by SV40 ST Is Mediated by a Cell Cycle-Regulated E2F Site. Virology 2005, 332, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Hackzell, A.; Wetterskog, D.; Ballági, A.; Izumi, H.; Funa, K. PRb, Myc and P53 Are Critically Involved in SV40 Large T Antigen Repression of PDGF β-Receptor Transcription. J. Cell Sci. 2004, 117, 3855–3865. [Google Scholar] [CrossRef][Green Version]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 Large T Antigen Targets Multiple Cellular Pathways to Elicit Cellular Transformation. Oncogene 2005, 7729–7745. [Google Scholar] [CrossRef]
- Sontag, E.; Fedorov, S.; Kamibayashi, C.; Robbins, D.; Cobb, M.; Mumby, M. The Interaction of SV40 Small Tumor Antigen with Protein Phosphatase 2A Stimulates the Map Kinase Pathway and Induces Cell Proliferation. Cell 1993, 75, 887–897. [Google Scholar] [CrossRef]
- Kotadia, S.; Kao, L.R.; Comerford, S.A.; Jones, R.T.; Hammer, R.E.; Megraw, T.L. PP2A-Dependent Disruption of Centrosome Replication and Cytoskeleton Organization in Drosophila by SV40 Small Tumor Antigen. Oncogene 2008, 27, 6334–6346. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Garriga, J.; Kurimchak, A.; Graña, X. Cyclin E and SV40 Small t Antigen Cooperate to Bypass Quiescence and Contribute to Transformation by Activating CDK2 in Human Fibroblasts. J. Biol. Chem. 2008, 283, 11280–11292. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Garriga, J.; Padgaonkar, A.; Kurimchak, A.; Cook, J.G.; Graña, X. Coordinated Activation of the Origin Licensing Factor CDC6 and CDK2 in Resting Human Fibroblasts Expressing SV40 Small T Antigen and Cyclin, E.J. Biol. Chem. 2009, 284, 14126–14135. [Google Scholar] [CrossRef]
- Kanca, O.; Bellen, H.J.; Schnorrer, F. Gene Tagging Strategies to Assess Protein Expression, Localization, and Function in Drosophila. Genetics 2017, 207, 389–412. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.T.; Sarrion-Perdigones, A.; Vandeventer, P.J.; Abel, N.S.; Christiansen, A.E.; Hoffman, K.L. Genome Engineering: Drosophila Melanogaster and Beyond. Wiley Interdiscip. Rev. Dev. Biol. 2016, 5, 233–267. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.; Demicheli, V.; Di Pietrantonj, C.; Rivetti, D. Amantadine and Rimantadine for Influenza A in Adults. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Wongsaroj, L.; Saninjuk, K.; Romsang, A.; Duang-nkern, J.; Trinachartvanit, W.; Vattanaviboon, P.; Mongkolsuk, S. Pseudomonas aeruginosa Glutathione Biosynthesis Genes Play Multiple Roles in Stress Protection, Bacterial Virulence and Biofilm Formation. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 697–708. [Google Scholar] [CrossRef]
- Shaw, M.L.; Stone, K.L.; Colangelo, C.M.; Gulcicek, E.E.; Palese, P. Cellular Proteins in Influenza Virus Particles. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.L.; Chohan, K.; Swenson, J.; Lajeunesse, D. A Drosophila Model for Genetic Analysis of Influenza Viral/Host Interactions. Genetics 2011, 189, 495–506. [Google Scholar] [CrossRef]
- Hughey, P.G.; Compans, R.W.; Zebedee, S.L.; Lamb, R.A. Expression of the Influenza A Virus M2 Protein Is Restricted to Apical Surfaces of Polarized Epithelial Cells. J. Virol. 1992, 66, 5542–5552. [Google Scholar] [CrossRef] [PubMed]
- Dow, J.A.T.; Davies, S.A.; Guo, Y.; Graham, S.; Finbow, M.E.; Kaiser, K. Molecular Genetic Analysis of V-ATPase Function in Drosophila Melanogaster. J. Exp. Biol. 1997, 200, 237–245. [Google Scholar] [PubMed]
- Niikura, K. Vacuolar ATPase as a Drug Discovery Target. Drug News Perspect. 2006, 139–144. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Mah, T.F.C.; O’Toole, G.A. Mechanisms of Biofilm Resistance to Antimicrobial Agents. Trends Microbiol. 2001, 34–39. [Google Scholar] [CrossRef]
- Furukawa, S.; Kuchma, S.L.; O’Toole, G.A. Keeping Their Options Open: Acute versus Persistent Infections. J. Bacteriol. 2006, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.Y.; Sim, N.; Cho, Y.H. Antibacterial Efficacy of Temperate Phage-Mediated Inhibition of Bacterial Group Motilities. Antimicrob. Agents Chemother. 2012, 56, 5612–5617. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, H.M.; McKean, K.A.; Wang, I.-N. Phage Fitness May Help Predict Phage Therapy Efficacy. Bacteriophage 2014, 4, e964081. [Google Scholar] [CrossRef]
- Heo, Y.J.; Lee, Y.R.; Jung, H.H.; Lee, J.; Ko, G.P.; Cho, Y.H. Antibacterial Efficacy of Phages against Pseudomonas aeruginosa Infections in Mice and Drosophila Melanogaster. Antimicrob. Agents Chemother. 2009, 53, 2469–2474. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sabin, L.R.; Hanna, S.L.; Cherry, S. Innate Antiviral Immunity in Drosophila. Curr. Opin. Immunol. 2010, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, M.; Kimura, A. Genomic View of the Evolution of the Complement System. Immunogenetics 2006, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Vlisidou, I.; Wood, W. Drosophila Blood Cells and Their Role in Immune Responses. FEBS J. 2015, 1368–1382. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Pathogens | Pathogenic Proteins | Section | Biological Processes Affected | In vivo Functions of Pathogenic Proteins |
|---|---|---|---|---|---|
| Bacteria | Aeromonas salmonicida | AopP | Section 2.2 | NF-kB signaling | Blocks the nuclear translocation of NF-kB (Relish and DIF), inhibiting both IMD and Toll pathways. |
| Section 2.2 | Apoptosis | Facilitates the cleavage of Caspase-3, inducing apoptosis. | |||
| Bacteria | Bacillus anthracis | EF | Section 4.3 | Hedgehog signaling | Hyperactivates PKA through its adenyl cyclase activity, activating Hedgehog signaling. Genetically interacts with hedgehog. |
| Section 4.3 | Notch signaling | Alters the subcellular localization of Delta ligands via affecting Rab11-dependent vesicle trafficking. Acts synergistically with LF protein. | |||
| Section 4.3 | Cell-cell adhesion | Alters the subcellular localization of E-Cadherin by activation of Epac through its adenyl cyclase activity. | |||
| LF | Section 4.4 | JNK signaling | Inhibits JNK signaling upstream of hep (JNKK) in the developing thorax. | ||
| Section 4.4 | EGFR signaling | Inhibits EGFR signaling in the developing wing disc through unknown mechanisms. Genetically interacts with Dsor1 (MAPKKK). | |||
| Section 4.4 | Notch signaling | Alters the subcellular localization of Delta ligand via affecting Sec15-dependent vesicle trafficking. Acts synergistically with EF protein. | |||
| Section 4.4 | Cell-cell adhesion | Alters the subcellular localization of E-Cadherin. | |||
| Bacteria | Helicobacter pylori | CagA | Section 4.6 | EGFR/Sevenless signaling | Activates EGFR signaling by mimicking the function of Dos (Gab-family protein) in a phosphorylation dependent manner through Corkscrew (SHP-2). |
| Section 4.6 | Cytoskeletal organization | Causes over-activation and altered subcellular localization of Spaghetti squash (Myosin light chain) via Rho GTPase in a phosphorylation-dependent manner. | |||
| Section 4.6 | JNK signaling and apoptosis | Activates JNK signaling upstream of Bsk (JNK), leading to increase in apoptosis. | |||
| Section 4.6 | Tumor metastasis | Synergizes with an oncogenic form of Ras (RasV12) to facilitate the invasion of tumors formed in the eye. Genetically interacts with basolateral protein coding genes dlg1 and l(2)gl that function as tumor suppressors. | |||
| Section 4.6 | Microbiome homeostasis | Causes dysbiosis of gastric microbiota when expressed in the digestive tract, leading to activation of immune responses in a phosphorylation-dependent manner. | |||
| Bacteria | Pseudomonas aeruginosa | ExoS | Section 3.1 | Phagocytosis | Inhibits phagocytosis by blocking Rac2 (Rho family GTPase) function in hemocytes. |
| gshA, gshB | Section 5.2 | Bacterial stress resistance and biofilm production | Protects bacteria from ROS while negatively regulating the formation of biofilms. | ||
| Bacteria | Salmonellae enterica | AvrA | Section 2.1 | NF-kB signaling | Blocks the nuclear translocation of NF-kB (Relish) in an enzymatic activity-dependent manner, inhibiting the IMD pathway. |
| Section 2.1 | JNK signaling | Decreases activity of MKK4 (JNKK), inhibiting JNK signaling. | |||
| Bacteria | Vibrio cholerae | CtxA | Section 4.4 | Notch signaling | Alters the subcellular localization of Delta ligand via affecting Rab11-dependent vesicle trafficking in an adenyl cyclase activity-dependent manner. |
| Section 4.4 | Cell-cell adhesion | Alters the subcellular localization of E-Cadherin by affecting Rab11-dependent vesicle trafficking. | |||
| Virus | Epstein Barr virus (EBV) | BRLF1 | Section 3.4 | Cell proliferation | Promotes cell proliferation. Genetically interacts with p53, Tor, reaper, and other genes. |
| BZLF1 | Section 3.4 | Apoptosis and cell proliferation | Works with shaven (Pax transcription factor) to facilitate apoptosis and inhibit cell proliferation. Genetically interacts with p53, Tor, reaper, and other genes. | ||
| Virus | Influenza virus | M2 | Section 5.1 | pH homeostasis | Increases intracellular pH through its function as a proton channel. |
| Virus | Human Cytomegalovirus (HCMV) | IE72, IE86 | Section 4.5 | Cell-cell adhesion | Alters the subcellular localization of Armadillo (b-Catenin). |
| Virus | Human Immunodeficiency Virus (HIV) | Nef | Section 2.5 | JNK signaling and apoptosis | Activates JNK signaling, leading to an increase in apoptosis. Genetically interacts with bsk (JNK) and hep (JNKK). |
| Tat | Section 4.8 | Cytoskeletal organization | Decreases the rate of Tubulin polymerization during cytoplasmic streaming during oogenesis and mitosis during early embryogenesis. | ||
| Section 4.8 | Protein translation | Interferes with ribosome biosynthesis by binding to pre-rRNA and Fibrillarin. | |||
| Vpu | Section 2.4 | NF-kB signaling | Inhibits Slmb (b-TRcP) in a phosphorylation-dependent manner, activating the Toll pathway. | ||
| Section 3.3 | JNK signaling and apoptosis | Activates JNK signaling upstream of hep (JNKK) in a phosphorylation-independent manner, leading to an increase in apoptosis. | |||
| Virus | Human papillomavirus (HPV) | E6 | Section 4.7 | Cell adhesion and polarity | Causes disruption of cell adhesion and polarity by degrading proteins such as Dlg1, Scrib, and Magi with Ube3A (E3 ligase) during wing development. |
| Section 4.7 | Insulin signaling | Genetically interacts with a dominant-negative form of Insulin receptor during eye development. | |||
| Section 4.7 | Epithelial-to-mesenchymal transition (EMT) | Genetically interacts with oncogenic forms of Ras and Notch to contribute to EMT. | |||
| Virus | Human T Cell Lymphotropic Virus type 1 (HTLV-1) | Tax1 | Section 2.3 | NF-kB signaling | Inhibits Kenny (IKKg) in a Urmylation-dependent manner, activating the IMD pathway. |
| Virus | Severe Acute Respiratory Syndrome Coronavirus-1 (SARS-CoV-1) | 3a | Section 3.2 | PKB/AKT signaling and apoptosis | Inhibits the Pdk1-Akt1 axis of the PKB/AKT pathway, leading to an increase in apoptosis. |
| M | Section 3.2 | Apoptosis | Promotes apoptosis in an ion channel activity-dependent fashion. | ||
| Virus | Simian vacuolating virus 40 (SV40) | Small T antigen | Section 4.9 | Mitosis | Causes mitotic spindle abnormalities by working with PP2A and upregulating Cyclin E expression. |
| Virus | Zika virus (ZIKV) | NS4A | Section 4.1 | Asymmetric cell division | Inhibits Ball (Vrk1) to misregulate proper segregation of cell polarity regulators in neural stem cells. |
| Section 4.1 | Apoptosis | Induces apoptosis in the nervous system. | |||
| Section 4.2 | JAK-STAT signaling | Inhibits JAK-STAT signaling downstream of hopscotch (JAK kinase) in the developing wing. | |||
| Section 4.2 | Notch signaling | Inhibits Notch signaling in the developing wing through unknown mechanisms. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harnish, J.M.; Link, N.; Yamamoto, S. Drosophila as a Model for Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 2724. https://doi.org/10.3390/ijms22052724
Harnish JM, Link N, Yamamoto S. Drosophila as a Model for Infectious Diseases. International Journal of Molecular Sciences. 2021; 22(5):2724. https://doi.org/10.3390/ijms22052724
Chicago/Turabian StyleHarnish, J. Michael, Nichole Link, and Shinya Yamamoto. 2021. "Drosophila as a Model for Infectious Diseases" International Journal of Molecular Sciences 22, no. 5: 2724. https://doi.org/10.3390/ijms22052724
APA StyleHarnish, J. M., Link, N., & Yamamoto, S. (2021). Drosophila as a Model for Infectious Diseases. International Journal of Molecular Sciences, 22(5), 2724. https://doi.org/10.3390/ijms22052724