
The Role of Immunogenetics in COVID-19

 ,
, 
 ,
, 

 and
and
Abstract
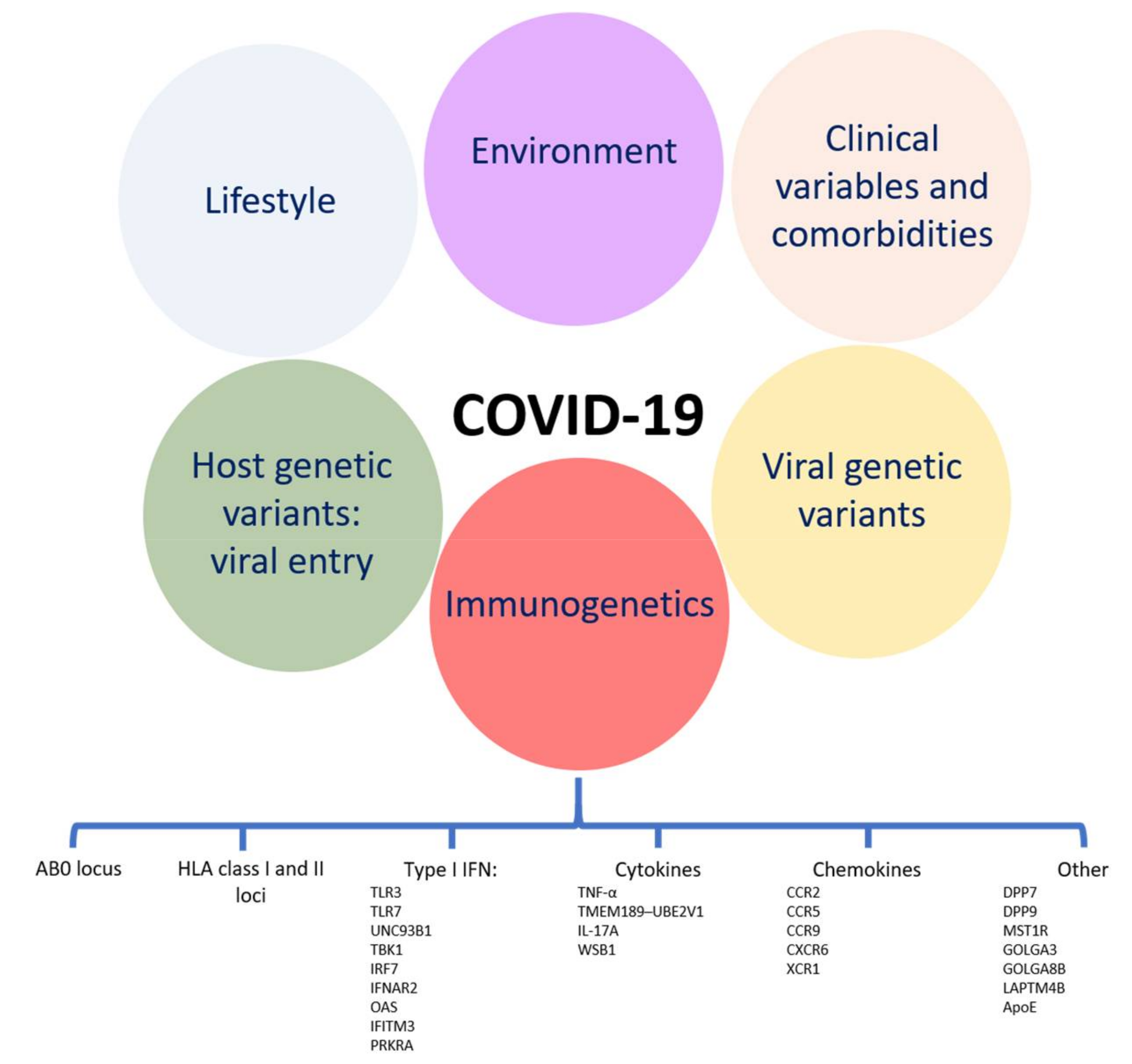
1. Introduction
2. AB0 Groups
3. HLA
3.1. Incidence and Susceptibility
3.2. Disease Severity
3.3. Mortality
3.4. Transplanted Patients
3.5. Functional and Mechanistic Considerations
4. Other Immune Response Genes
4.1. Type I Interferons and Players of Their Molecular Pathways
4.2. Other Cytokines, Chemokines and Their Signaling Pathways
4.3. Other Genetic Determinants of Establishment/Maintenance/Resolution of the Immune Response and Antigen Presentation
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| AD | Autosomal-dominant |
| AH | Ancestral haplotype |
| AIDS | Acquired immunodeficiency syndrome |
| APACHE | Acute Physiology and Chronic Health Evaluation |
| ApoE | Apoliprotein E |
| AR | Autosomal-recessive |
| ARDS | Acute respiratory distress syndrome |
| CD40L | CD40 ligand |
| CDC | Centers for Disease Control and Prevention |
| CCR2 | C-C Motif Chemokine Receptor 2 |
| CCR3 | C-C Motif Chemokine Receptor 3 |
| CCR5 | C-C Motif Chemokine Receptor 5 |
| CCR9 | C-C Motif Chemokine Receptor 9 |
| COVID-19 | Coronavirus disease 19 |
| CRP | C-reactive protein |
| CXCR3 | C-X-C Motif Chemokine Receptor 3 |
| CXCR6 | C-X-C Motif Chemokine Receptor 6 |
| DPP | Dipeptidyl peptidase |
| EBV | Epstein-Barr Virus |
| ECMO | Extracorporeal membrane oxygenation |
| eQTL | Expression quantitative locus |
| FOXP3 | Forkhead box p3 |
| GM | Immunoglobulin G heavy chain |
| GOLGA3 | Golgin subfamily A 3 |
| GOLGA8B | Golgin A8 family member B |
| GWAS | Genome wide association study |
| HIV | Human immunodeficiency virus |
| HLA | Human Leukocyte Antigens |
| ICU | Intensive care unit |
| IFITM3 | Interferon-induced transmembrane protein 3 |
| IFNB1 | Interferon β 1 |
| IFN-α | Interferon-α |
| IFN-β | Interferon-β |
| IFN-γ | Interferon-γ |
| IFNAR1 | IFN-α receptor 1 |
| IFNAR2 | IFN-α receptor 2 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| IL-10 | Interleukin-10 |
| IL-17A | Interleukin-17A |
| IL-17F | Interleukin-17F |
| IL-21 | Interleukin-21 |
| IP-10 | Interferon γ-induced protein-10 |
| IRF | IFN regulatory factors |
| ISG15 | Ubiquitin-like modifier ISG15 |
| KIR | Killer Immunoglobulin Receptors |
| KIR2DL2 | Killer Cell Immunoglobulin-Like Receptor 2DL2 |
| KIR2DL3 | Killer Cell Immunoglobulin-Like Receptor 2DL3 |
| LAPTM4B | Lysosomal Protein Transmembrane 4 β |
| MIP-1α | Macrophage inflammatory protein 1α |
| MIP-1β | Macrophage inflammatory protein 1β |
| MMR | Measles, mumps, and rubella |
| MST1R | Macrophage stimulating 1 receptor |
| NEMO/IKBKG | NF-κB essential modulator |
| NK | Natural Killer |
| OAS | 2’-5’-Oligoadenylate synthetase |
| PHA | Phytohemagglutinin |
| PKR | IFN-induced, double-stranded RNA-activated protein kinase |
| pLOF | Predicted to be loss-of-function |
| PRKRA | Protein Activator of The Interferon-Induced Protein Kinase |
| RBC | Red blood cells |
| SNP | Single nucleotide polymorphism |
| SOFA | Sepsis-related Organ Failure Assessment |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT2 | Signal transducer and activator of transcription 2 |
| TBK1 | TANK binding kinase 1 |
| TICAM1/TRIF | TIR-domain containing adaptor inducing IFN-β |
| TLR | Toll like receptor |
| TMEM189 | Transmembrane protein 189 |
| TMPRSS2 | Transmembrane protease serine protease 2 |
| TNF-α | Tumor necrosis factor-α |
| TRAF3 | TNF Receptor Associated Factor 3 |
| UBE2V1 | Ubiquitin-conjugating enzyme E2 variant 1 |
| UNC93B1 | Unc-93 homolog B1 |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
| VWF | von Willebrand factor |
| WSB1 | WD Repeat And SOCS Box Containing 1 |
| XCL1 | X-C Motif Chemokine Ligand 1 |
| XCR1 | X-C Motif Chemokine Receptor 1 |
References
- Guo, G.; Ye, L.; Pan, K.; Chen, Y.; Xing, D.; Yan, K.; Chen, Z.; Ding, N.; Li, W.; Huang, H.; et al. New Insights of Emerging SARS-CoV-2: Epidemiology, Etiology, Clinical Features, Clinical Treatment, and Prevention. Front. Cell Dev. Biol. 2020, 410. PMCID: PMC7256189. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14, PMCID: PMC7537588. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2020. PMCID: PMC7361342. [Google Scholar] [CrossRef] [PubMed]
- Tizaoui, K.; Zidi, I.; Lee, K.H.; Ghayda, R.A.; Hong, S.H.; Li, H.; Smith, L.; Koyanagi, A.; Jacob, L.; Kronbichler, A.; et al. Update of the current knowledge on genetics, evolution, immunopathogenesis, and transmission for coronavirus disease 19 (COVID-19). Int. J. Biol. Sci. 2020, 16, 2906–2923, PMCID: PMC7545713. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Hilgenfeld, R.; Zabel, P. Molecular mechanisms of severe acute respiratory syndrome (SARS). Respir. Res. 2005, 6, 8, PMCID: PMC548145. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Poutanen, S.M.; Low, D.E.; Lode, H.; Welte, T.; Zabel, P. Treatment and vaccines for severe acute respiratory syndrome. Lancet Infect. Dis. 2005, 5, 147–155, PMCID:PMC7106466. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Tabarsi, P.; Varahram, M.; Folkerts, G.; Adcock, I.M. The Immune Response and Immunopathology of COVID-19. Front. Immunol. 2020, 11, 2037, PMCID: PMC7479965. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, S. Understand variability of COVID-19 through population and tissue variations in expression of SARS-CoV-2 host genes. Inf. Med. Unlocked 2020, 21, 100443, PMCID: PMC7550072. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Bandopadhyay, A.; Das, D.; Pandey, R.K.; Singh, V.; Khanam, N.; Srivastava, N.; Singh, P.P.; Dubey, P.K.; Pathak, A.; et al. Genetic Association of ACE2 rs2285666 Polymorphism With COVID-19 Spatial Distribution in India. Front. Genet. 2020, 11, 564741, PMCID: PMC7545580. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef]
- Barash, A.; Machluf, Y.; Ariel, I.; Dekel, Y. The Pursuit of COVID-19 Biomarkers: Putting the Spotlight on ACE2 and TMPRSS2 Regulatory Sequences. Front. Med. 2020, 7, 582793, PMCID: PMC7661736. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Sreenivasulu, K.; Mitra, P.; Misra, S.; Sharma, P. Role of Genetic Variants and Gene Expression in the Susceptibility and Severity of COVID-19. Ann. Lab. Med. 2021, 41, 129–138, PMCID: PMC7591285. [Google Scholar] [CrossRef] [PubMed]
- Chiappelli, F. CoViD-19 Susceptibility. Bioinformation 2020, 16, 501–504, PMCID: PMC7505245. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 40, PMCID: PMC7578581. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469, PMCID: PMC7477538. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Carrington, M. Immunogenetics of viral infections. Curr. Opin. Immunol. 2005, 17, 510–516. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.; Peterson, A.; Tian, Y.; Sang, Y. Immunogenetic Association Underlying Severe COVID-19. Vaccines 2020, 8, 700. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Caruso, C.; Vasto, S. Possible role of ABO system in age-related diseases and longevity: A narrative review. Immun. Ageing 2014, 11, 16, PMCID: PMC4265994. [Google Scholar] [CrossRef] [PubMed]
- Vasto, S.; Caruso, C.; Castiglia, L.; Duro, G.; Monastero, R.; Rizzo, C. Blood group does not appear to affect longevity a pilot study in centenarians from Western Sicily. Biogerontology 2011, 12, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. Clin. Infect. Dis. 2020, ciaa1150, PMCID: PMC7454371. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A. Genetic Risk of Severe Covid-19. N. Engl. J. Med. 2020, 383, 1590–1591, PMCID: PMC7583681. [Google Scholar] [CrossRef] [PubMed]
- Zietz, M.; Zucker, J.; Tatonetti, N.P. Associations between blood type and COVID-19 infection, intubation, and death. Nat. Commun. 2020, 11, 5761, PMCID: PMC7666188. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, W.; Li, B.; Li, D.J.; Zhang, J.; Zhao, F. Association between ABO Blood Group System and COVID-19 Susceptibility in Wuhan. Front. Cell Infect. Microbiol. 2020, 10, 404, PMCID: PMC7385064. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-Diaz, E.; Llopis, J.; Parra, R.; Roig, I.; Ferrer, G.; Grifols, J.; Millán, A.; Ene, G.; Ramiro, L.; Maglio, L.; et al. Relationship between the ABO blood group and COVID-19 susceptibility, severity and mortality in two cohorts of patients. Blood Transfus. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.B.; Gu, D.Z.; Yu, J.N.; Yang, J.; Shen, W.Q. Association between ABO blood groups and COVID-19 infection, severity and demise: A systematic review and meta-analysis. Infect. Genet. Evol. 2020, 84, 104485, PMCID: PMC7391292. [Google Scholar] [CrossRef] [PubMed]
- Barnkob, M.B.; Pottegård, A.; Støvring, H.; Haunstrup, T.M.; Homburg, K.; Larsen, R.; Hansen, M.B.; Titlestad, K.; Aagaard, B.; Møller, B.K.; et al. Reduced prevalence of SARS-CoV-2 infection in ABO blood group O. Blood Adv. 2020, 4, 4990–4993, PMCID: PMC7594382. [Google Scholar] [CrossRef] [PubMed]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534, PMCID: PMC7315890. [Google Scholar] [CrossRef]
- Amoroso, A.; Magistroni, P.; Vespasiano, F.; Bella, A.; Bellino, S.; Puoti, F.; Alizzi, S.; Vaisitti, T.; Boros, S.; Grossi, P.A.; et al. HLA and AB0 Polymorphisms May Influence SARS-CoV-2 Infection and COVID-19 Severity. Transplantation 2020. [Google Scholar] [CrossRef] [PubMed]
- Latz, C.A.; DeCarlo, C.; Boitano, L.; Png, C.Y.M.; Patell, R.; Conrad, M.F.; Eagleton, M.; Dua, A. Blood type and outcomes in patients with COVID-19. Ann. Hematol. 2020, 99, 2113–2118, PMCID: PMC7354354. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Albaiceta, G.M.; Clemente, M.G.; Gómez, J. Lack of association between SNPsrs8176719(O blood group) and COVID-19: Data from Spanish age matched patients and controls. Transfusion 2020. [Google Scholar] [CrossRef] [PubMed]
- Licastro, F.; Candore, G.; Lio, D.; Porcellini, E.; Colonna-Romano, G.; Franceschi, C.; Caruso, C. Innate immunity and inflammation in ageing: A key for understanding age-related diseases. Immun. Ageing 2005, 2, 8, PMCID: PMC1166571. [Google Scholar] [CrossRef] [PubMed]
- Lio, D.; Scola, L.; Giarratana, R.M.; Candore, G.; Colonna-Romano, G.; Caruso, C.; Balistreri, C.R. SARS CoV2 infection _The longevity study perspectives. Ageing Res. Rev. 2021, 67, 101299, PMCID: PMC7885677. [Google Scholar] [CrossRef] [PubMed]
- Cooling, L. Blood Groups in Infection and Host Susceptibility. Clin. Microbiol. Rev. 2015, 28, 801–870, PMCID: PMC4475644. [Google Scholar] [CrossRef] [PubMed]
- Liumbruno, G.M.; Franchini, M. Beyond immunohaematology: The role of the ABO blood group in human diseases. Blood Transfus. 2013, 11, 491–499, PMCID: PMC3827391. [Google Scholar] [CrossRef] [PubMed]
- Handunnetthi, L.; Ramagopalan, S.V.; Ebers, G.C.; Knight, J.C. Regulation of major histocompatibility complex class II gene expression, genetic variation and disease. Genes Immun. 2010, 11, 99–112, PMCID: PMC2987717. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Yang, G.-y.; Song, Y.; Zhao, X.; So, C.; Liao, J.; Wang, L.-D.; Yang, C.S. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis 2001, 22, 1615–1623. [Google Scholar] [CrossRef]
- Singh, S.; Banerjee, S. Downregulation of HLA-ABC expression through promoter hypermethylation and downmodulation of MIC-A/B surface expression in LMP2A-positive epithelial carcinoma cell lines. Sci. Rep. 2020, 10, 5415. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Mutti, L.; Pentimalli, F.; Baglio, G.; Saladino, R.E.; Sileri, P.; Giordano, A. HLA-B*44 and C*01 Prevalence Correlates with Covid19 Spreading across Italy. Int. J. Mol. Sci. 2020, 21, 5205, PMCID: PMC7432860. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Franco, A.; Barrios, Y.; Cáceres, J.J.; Solé-Violán, J.; Perez, A.; Marcos, Y.; Ramos, J.A.; Ramos-Gómez, L.; et al. HLA genetic polymorphisms and prognosis of patients with COVID-19. Med. Intensiva. 2021, 45, 96–103, PMCID: PMC7474921. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Andreani, M.; Biancolella, M.; Liberatoscioli, L.; Passarelli, C.; Colona, V.L.; Rogliani, P.; Leonardis, F.; Campana, A.; Carsetti, R.; et al. HLA allele frequencies and susceptibility to COVID-19 in a group of 99 Italian patients. HLA 2020, 96, 610–614, PMCID: PMC7461491. [Google Scholar] [CrossRef] [PubMed]
- Kachuri, L.; Francis, S.S.; Morrison, M.; Wendt, G.A.; Bossé, Y.; Cavazos, T.B.; Rashkin, S.R.; Ziv, E.; Witte, J.S. The landscape of host genetic factors involved in infection to common viruses and SARS-CoV-2. medRxiv 2020, 12, 93, Update in Genome Med. 2020, 12, 93. PMCID: PMC7273301. [Google Scholar] [CrossRef]
- Benlyamani, I.; Venet, F.; Coudereau, R.; Gossez, M.; Monneret, G. Monocyte HLA-DR Measurement by Flow Cytometry in COVID-19 Patients: An Interim Review. Cytom. A 2020. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Pittet, J.F. Human Leukocyte Antigen-DR Deficiency and Immunosuppression-Related End-Organ Failure in SARS-CoV2 Infection. Anesth. Analg. 2020, 131, 989–992, PMCID: PMC7386673. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Zhang, J.; He, J.; Zhu, F. Distribution of HLA allele frequencies in 82 Chinese individuals with coronavirus disease-2019 (COVID-19). HLA 2020, 96, 194–196, PMCID: PMC7276866. [Google Scholar] [CrossRef]
- Yung, Y.L.; Cheng, C.K.; Chan, H.Y.; Xia, J.T.; Lau, K.M.; Wong, R.S.M.; Wu, A.K.L.; Chu, R.W.; Wong, A.C.C.; Chow, E.Y.D.; et al. Association of HLA-B22 serotype with SARS-CoV-2 susceptibility in Hong Kong Chinese patients. HLA 2020. [Google Scholar] [CrossRef] [PubMed]
- Poulton, K.; Wright, P.; Hughes, P.; Savic, S.; Welberry Smith, M.; Guiver, M.; Morton, M.; van Dellen, D.; Tholouli, E.; Wynn, R.; et al. A role for human leucocyte antigens in the susceptibility to SARS-Cov-2 infection observed in transplant patients. Int. J. Immunogenet. 2020, 47, 324–328, PMCID: PMC7361549. [Google Scholar] [CrossRef]
- Rosenbaum, J.T.; Hamilton, H.; Weisman, M.H.; Reveille, J.D.; Winthrop, K.L.; Choi, D. The Effect of HLA-B27 on Susceptibility and Severity of COVID-19. J. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KIR2DL2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KIR2DL3&keywords=KIR2DL3 (accessed on 26 February 2021).
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00510-20, PMCID: PMC7307149. [Google Scholar] [CrossRef] [PubMed]
- Olwenyi, O.A.; Dyavar, S.R.; Acharya, A.; Podany, A.T.; Fletcher, C.V.; Ng, C.L.; Reid, S.P.; Byrareddy, S.N. Immuno-epidemiology and pathophysiology of coronavirus disease 2019 (COVID-19). J. Mol. Med. 2020, 98, 1369–1383, PMCID: PMC7431311. [Google Scholar] [CrossRef]
- Tomita, Y.; Ikeda, T.; Sato, R.; Sakagami, T. Association between HLA gene polymorphisms and mortality of COVID-19: An in silico analysis. Immun. Inflamm. Dis. 2020, 8, 684–694, PMCID: PMC7654404. [Google Scholar] [CrossRef]
- Iturrieta-Zuazo, I.; Rita, C.G.; García-Soidán, A.; de Malet Pintos-Fonseca, A.; Alonso-Alarcón, N.; Pariente-Rodríguez, R.; Tejeda-Velarde, A.; Serrano-Villar, S.; Castañer-Alabau, J.L.; Nieto-Gañán, I. Possible role of HLA class-I genotype in SARS-CoV-2 infection and progression: A pilot study in a cohort of Covid-19 Spanish patients. Clin. Immunol. 2020, 219, 108572, PMCID: PMC7428760. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, J.P.; Carnalla-Cortés, M.; Pacheco-Olvera, D.L.; Ocampo-Godínez, J.M.; Oliva-Ramírez, J.; Moreno-Manjón, J.; Bernal-Alferes, B.; López-Olmedo, N.; García-Latorre, E.; Domínguez-López, M.L.; et al. A bioinformatic prediction of antigen presentation from SARS-CoV-2 spike protein revealed a theoretical correlation of HLA-DRB1*01 with COVID-19 fatality in Mexican population: An ecological approach. J. Med. Virol. 2020. PMCID: PMC7537233. [Google Scholar] [CrossRef]
- Pisanti, S.; Deelen, J.; Gallina, A.M.; Caputo, M.; Citro, M.; Abate, M.; Sacchi, N.; Vecchione, C.; Martinelli, R. Correlation of the two most frequent HLA haplotypes in the Italian population to the differential regional incidence of Covid-19. J. Transl. Med. 2020, 18, 352, PMCID: PMC7491019. [Google Scholar] [CrossRef] [PubMed]
- Gambino, C.M.; Aiello, A.; Accardi, G.; Caruso, C.; Candore, G. Autoimmune diseases and 8.1 ancestral haplotype: An update. HLA 2018, 92, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Habel, J.R.; Nguyen, T.H.O.; van de Sandt, C.E.; Juno, J.A.; Chaurasia, P.; Wragg, K.; Koutsakos, M.; Hensen, L.; Jia, X.; Chua, B.; et al. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A*02:01 phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 24384–24391, PMCID: PMC7533701. [Google Scholar] [CrossRef]
- Caruso, C.; Candore, G.; Romano, G.C.; Lio, D.; Bonafè, M.; Valensin, S.; Franceschi, C. Immunogenetics of longevity. Is major histocompatibility complex polymorphism relevant to the control of human longevity? A review of literature data. Mech. Ageing Dev. 2001, 122, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Player, M.A.; Barracchini, K.C.; Simonis, T.B.; Rivoltini, L.; Arienti, F.; Castelli, C.; Mazzocchi, A.; Belli, F.; Parmiani, G.; Marincola, F.M. Differences in frequency distribution of HLA-A2 subtypes between North American and Italian white melanoma patients: Relevance for epitope specific vaccination protocols. J. Immunother. Emphas. Tumor Immunol. 1996, 19, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Accardi, G.; Caruso, C. Genetic variation in human leukocyte antigen and susceptibility to acute myeloid leukemia. Acta Haematol. 2015, 133, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.A.; Sullivan, L.C.; Ottenhoff, T.H. Characteristics of HLA-E Restricted T-Cell Responses and Their Role in Infectious Diseases. J. Immunol. Res. 2016, 2016, 2695396, PMCID: PMC5028793. [Google Scholar] [CrossRef]
- Kanevskiy, L.; Erokhina, S.; Kobyzeva, P.; Streltsova, M.; Sapozhnikov, A.; Kovalenko, E. Dimorphism of HLA-E and its Disease Association. Int. J. Mol. Sci. 2019, 20, 5496, PMCID: PMC6862560. [Google Scholar] [CrossRef]
- Lopez, L.; Sang, P.C.; Tian, Y.; Sang, Y. Dysregulated Interferon Response Underlying Severe COVID-19. Viruses 2020, 12, 1433. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G. The Role of Type I Interferons in the Pathogenesis and Treatment of COVID-19. Front. Immunol. 2020, 11, 595739, PMCID: PMC7561359. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2020, 65, 101205, PMCID: PMC7604159. [Google Scholar] [CrossRef] [PubMed]
- Angioni, R.; Sánchez-Rodríguez, R.; Munari, F.; Bertoldi, N.; Arcidiacono, D.; Cavinato, S.; Marturano, D.; Zaramella, A.; Realdon, S.; Cattelan, A.; et al. Age- severity matched cytokine profiling reveals specific signatures in Covid-19 patients. Cell Death Dis. 2020, 11, 957, PMCID: PMC7646225. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. Immunesenescence: A Predisposing Risk Factor for the Development of COVID-19? Front. Immunol. 2020, 11, 573662, PMCID: PMC7573102. [Google Scholar] [CrossRef]
- Pietrobon, A.J.; Teixeira, F.M.E.; Sato, M.N. Immunosenescence and Inflammaging: Risk Factors of Severe COVID-19 in Older People. Front. Immunol. 2020, 11, 579220, PMCID: PMC7656138. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch. Immunol. Ther. Exp. 2016, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247, PMCID: PMC6773825. [Google Scholar] [CrossRef] [PubMed]
- Bouadma, L.; Wiedemann, A.; Patrier, J.; Surénaud, M.; Wicky, P.H.; Foucat, E.; Diehl, J.L.; Hejblum, B.P.; Sinnah, F.; de Montmollin, E.; et al. Immune Alterations in a Patient with SARS-CoV-2-Related Acute Respiratory Distress Syndrome. J. Clin. Immunol. 2020, 40, 1082–1092, PMCID: PMC7443154. [Google Scholar] [CrossRef] [PubMed]
- Jurado, A.; Martín, M.C.; Abad-Molina, C.; Orduña, A.; Martínez, A.; Ocaña, E.; Yarce, O.; Navas, A.M.; Trujillo, A.; Fernández, L.; et al. COVID-19: Age, Interleukin-6, C-reactive protein, and lymphocytes as key clues from a multicentre retrospective study. Immun Ageing. 2020, 17, 22, PMCID: PMC7426672. [Google Scholar] [CrossRef]
- Files, J.K.; Boppana, S.; Perez, M.D.; Sarkar, S.; Lowman, K.E.; Qin, K.; Sterrett, S.; Carlin, E.; Bansal, A.; Sabbaj, S.; et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J. Clin. Investig. 2020, 140491. [Google Scholar] [CrossRef] [PubMed]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020, 5, eabd6832, PMCID: PMC7665314. [Google Scholar] [CrossRef]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Life Sci. 2020, 258, 118167, PMCID: PMC7387997. [Google Scholar] [CrossRef] [PubMed]
- Cacciapuoti, S.; De Rosa, A.; Gelzo, M.; Megna, M.; Raia, M.; Pinchera, B.; Pontarelli, A.; Scotto, R.; Scala, E.; Scarano, F.; et al. Immunocytometric analysis of COVID patients: A contribution to personalized therapy? Life Sci. 2020, 261, 118355, PMCID: PMC7456265. [Google Scholar] [CrossRef]
- Cantenys-Molina, S.; Fernández-Cruz, E.; Francos, P.; Lopez Bernaldo de Quirós, J.C.; Muñoz, P.; Gil-Herrera, J. Lymphocyte subsets early predict mortality in a large series of hospitalized COVID-19 patients in Spain. Clin. Exp. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, W.; Dong, Y.; Wang, X.; Dai, D.; Liu, X.; Wu, Y.; Li, M.; Zhang, W.; Zhou, H.; et al. Elevated Exhaustion Levels of NK and CD8+ T Cells as Indicators for Progression and Prognosis of COVID-19 Disease. Front. Immunol. 2020, 11, 580237, PMCID: PMC7591707. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Luo, M.; Liu, J.; Wu, L.; Lin, X.; Li, R.; Wang, Z.; Zhong, H.; Zheng, W.; et al. Lymphopenia predicted illness severity and recovery in patients with COVID-19: A single-center, retrospective study. PLoS ONE 2020, 15, e0241659, PMCID: PMC7673513. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, Z.; Yin, Y.; Zhao, Y.; Tao, P.; Zhong, P. Association of Peripheral Lymphocyte and the Subset Levels With the Progression and Mortality of COVID-19: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7, 558545, PMCID: PMC7546210. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.; Faridi, R.M.; Sligl, W.; Shabani-Rad, M.T.; Dharmani-Khan, P.; Parker, A.; Kalra, A.; Tripathi, M.B.; Storek, J.; Cohen Tervaert, J.W.; et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. 2020, 4, 5035–5039, PMCID: PMC7594380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Liu, J.; Chen, L.; Zhou, F.; Jin, T.; Jiang, L.; Li, X.; Yang, M.; Wang, H. The characteristics and predictive role of lymphocyte subsets in COVID-19 patients. Int. J. Infect. Dis. 2020, 99, 92–99, PMCID: PMC7398035. [Google Scholar] [CrossRef]
- Zingaropoli, M.A.; Perri, V.; Pasculli, P.; Dezza, F.C.; Nijhawan, P.; Savelloni, G.; La Torre, G.; D’Agostino, C.; Mengoni, F.; Lichtner, M.; et al. Major reduction of NKT cells in patients with severe COVID-19 pneumonia. Clin. Immunol. 2020, 222, 108630, PMCID: PMC7661928. [Google Scholar] [CrossRef]
- Naumova, E.; Ivanova, M.; Pawelec, G. Immunogenetics of ageing. Int. J. Immunogenet. 2011, 38, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.D.; Conneely, K.N. The role of DNA methylation and hydroxymethylation in immunosenescence. Ageing Res. Rev. 2019, 51, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Allan, R.S. Epigenomic drivers of immune dysfunction in aging. Aging Cell 2019, 18, e12878, PMCID: PMC6351880. [Google Scholar] [CrossRef]
- Ostan, R.; Bucci, L.; Capri, M.; Salvioli, S.; Scurti, M.; Pini, E.; Monti, D.; Franceschi, C. Immunosenescence and immunogenetics of human longevity. Neuroimmunomodulation 2008, 15, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Atlante, S.; Mongelli, A.; Barbi, V.; Martelli, F.; Farsetti, A.; Gaetano, C. The epigenetic implication in coronavirus infection and therapy. Clin. Epigenet. 2020, 12, 156, PMCID: PMC7576975. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Kinoshita, N.; Ide, S.; Nomoto, H.; Nakamoto, T.; Saito, S.; Ishikane, M.; Kutsuna, S.; Hayakawa, K.; Hashimoto, M.; et al. Serum CCL17 level becomes a predictive marker to distinguish between mild/moderate and severe/critical disease in patients with COVID-19. Gene 2021, 766, 145145, PMCID: PMC7489253. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362, Erratum in 2020, 20, 448, doi:10.1038/s41577-020-0353-y. PMCID: PMC7201395. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Moosavi, M. Cytokine-targeted therapy in severely ill COVID-19 patients: Options and cautions. EJMO 2020, 4, 179–181. [Google Scholar] [CrossRef]
- Neumann, J.; Prezzemolo, T.; Vanderbeke, L.; Roca, C.P.; Gerbaux, M.; Janssens, S.; Willemsen, M.; Burton, O.; Van Mol, P.; Van Herck, Y.; et al. Increased IL-10-producing regulatory T cells are characteristic of severe cases of COVID-19. Clin. Transl. Immunol. 2020, 9, e1204, PMCID: PMC7662088. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb Pathog. 2021, 18, 104799, PMCID: PMC7889464. [Google Scholar] [CrossRef]
- Pontali, E.; Volpi, S.; Signori, A.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Montale, A.; Bustaffa, M.; Angelelli, A.; Caorsi, R.; et al. Efficacy of early anti-inflammatory treatment with high doses IV Anakinra with or without glucocorticoids in patients with severe COVID-19 pneumonia. J. Allergy Clin. Immunol. 2021. PMCID: PMC7865089. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Savale, L.; Dossier, A.; Ghosn, J.; Taillé, C.; Visseaux, B.; Jebreen, K.; Diallo, A.; Tesmoingt, C.; Morer, L.; et al. Glucocorticoids with low-dose anti-IL1 anakinra rescue in severe non-ICU COVID-19 infection: A cohort study. PLoS ONE 2020, 15, e0243961, PMCID: PMC7743937. [Google Scholar] [CrossRef]
- Bozzi, G.; Mangioni, D.; Minoia, F.; Aliberti, S.; Grasselli, G.; Barbetta, L.; Castelli, V.; Palomba, E.; Alagna, L.; Lombardi, A.; et al. Anakinra combined with methylprednisolone in patients with severe COVID-19 pneumonia and hyperinflammation: An observational cohort study. J. Allergy Clin. Immunol. 2021, 147, 561–566.e4, PMCID: PMC7674131. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Stewart, I.; Fabbri, L.; Moss, S.; Robinson, K.; Smyth, A.R.; Jenkins, G. Systematic review and meta-analysis of anakinra, sarilumab, siltuximab and tocilizumab for COVID-19. Thorax 2021. [Google Scholar] [CrossRef]
- The REMAP-CAP Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19—Preliminary report. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.01.07.21249390v1 (accessed on 26 February 2021). [CrossRef]
- Lauder, S.N.; Jones, E.; Smart, K.; Bloom, A.; Williams, A.S.; Hindley, J.P.; Ondondo, B.; Taylor, P.R.; Clement, M.; Fielding, C.; et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 2013, 43, 2613–2625, PMCID: PMC3886386. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456, PMCID: PMC7719805. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2020, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, M.; Manet, C.; Montagutelli, X. Host genetic susceptibility to viral infections: The role of type I interferon induction. Genes Immun. 2020, 1–15, PMCID: PMC7677911. [Google Scholar] [CrossRef]
- Lee, B.L.; Barton, G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014, 24, 360–369, PMCID: PMC4037363. [Google Scholar] [CrossRef]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFNAR1&keywords=IFNAR1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFNAR2&keywords=IFNAR2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=STAT1&keywords=stat1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=STAT2&keywords=stat2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TICAM1&keywords=TICAM1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TBK1&keywords=TBK1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TRAF3&keywords=traf3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=UNC93B1&keywords=UNC93B1 (accessed on 26 February 2021).
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IKBKG&keywords=nemo (accessed on 26 February 2021).
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020, 289, 198163, PMCID: PMC7480444. [Google Scholar] [CrossRef] [PubMed]
- Di Maria, E.; Latini, A.; Borgiani, P.; Novelli, G. Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): Rapid systematic review and field synopsis. Hum. Genom. 2020, 14, 30, PMCID: PMC7484929. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in Covid-19. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qin, L.; Zhao, Y.; Zhang, P.; Xu, B.; Li, K.; Liang, L.; Zhang, C.; Dai, Y.; Feng, Y.; et al. Interferon-Induced Transmembrane Protein 3 Genetic Variant rs12252-C Associated With Disease Severity in Coronavirus Disease 2019. J. Infect. Dis. 2020, 222, 34–37, PMCID: PMC7197559. [Google Scholar] [CrossRef]
- Gómez, J.; Albaiceta, G.M.; Cuesta-Llavona, E.; García-Clemente, M.; López-Larrea, C.; Amado-Rodríguez, L.; López-Alonso, I.; Melón, S.; Alvarez-Argüelles, M.E.; Gil-Peña, H.; et al. The Interferon-induced transmembrane protein 3 gene (IFITM3) rs12252 C variant is associated with COVID-19. Cytokine 2020, 137, 155354. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Giliberti, A.; Emiliozzi, A.; Valentino, F.; Bergantini, L.; Fallerini, C.; Anedda, F.; Amitrano, S.; Conticini, E.; Tita, R.; et al. Clinical and molecular characterization of COVID-19 hospitalized patients. PLoS ONE 2020, 15, e0242534, PMCID: PMC7673557. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Sultan, A.; Elashry, M.A.; Farag, A.; Mortada, M.I.; Ghannam, M.A.; Saed, A.M.; Ghoneem, S. Association of TNF-α G-308 a Promoter Polymorphism with the Course and Outcome of COVID-19 Patients. Immunol. Investig. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232, PMCID: PMC7314139. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. ApoE e4e4 Genotype and Mortality With COVID-19 in UK Biobank. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1801–1803, PMCID: PMC7337688. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.L.; Conn, G.L. RNA regulation of the antiviral protein 2′-5′-oligoadenylate synthetase. Wiley Interdiscip. Rev. RNA 2019, 10, e1534, PMCID: PMC6585406. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.omim.org/entry/605579?search=ifitm3&highlight=ifitm3 (accessed on 26 February 2021).
- Wellington, D.; Laurenson-Schafer, H.; Abdel-Haq, A.; Dong, T. IFITM3: How genetics influence influenza infection demographically. Biomed. J. 2019, 42, 19–26, PMCID: PMC6468115. [Google Scholar] [CrossRef]
- Lee, J.; Robinson, M.E.; Ma, N.; Artadji, D.; Ahmed, M.A.; Xiao, G.; Sadras, T.; Deb, G.; Winchester, J.; Cosgun, K.N.; et al. IFITM3 functions as a PIP3 scaffold to amplify PI3K signalling in B cells. Nature 2020, 588, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.G.; O’Keeffe, M.; Reading, P.C.; Wakim, L.M. Rapid interferon independent expression of IFITM3 following T cell activation protects cells from influenza virus infection. PLoS ONE 2019, 14, e0210132, PMCID: PMC6334895. [Google Scholar] [CrossRef]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455, PMCID: PMC7095036. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/603424?search=PRKRA&highlight=prkra (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=PRKRA&keywords=PRKRA (accessed on 26 February 2021).
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390, PMCID: PMC1170771. [Google Scholar] [CrossRef]
- Al-Meghaiseeb, E.S.; Al-Robayan, A.A.; Al-Otaibi, M.M.; Arfin, M.; Al-Asmari, A.K. Association of tumor necrosis factor-α and -β gene polymorphisms in inflammatory bowel disease. J. Inflamm. Res. 2016, 9, 133–140, PMCID: PMC4918894. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Cui, X.; Ning, L.; Wei, D. The effects of tumor necrosis factor-α (TNF-α) rs1800629 and rs361525 polymorphisms on sepsis risk. Oncotarget 2017, 8, 111456–111469, PMCID: PMC5762335. [Google Scholar] [CrossRef]
- Shi, C.; Zhao, H. Association between Tumor Necrosis Factor-308 G/A Polymorphism and Chronic Obstructive Pulmonary Disease Risk in Chinese Population: Evidence from a Meta-Analysis. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.K.; Han, Y.R.; Hong, D.; Chon, J.; Chung, J.H.; Hong, S.J.; Park, M.S.; Ban, J.Y. Promoter Polymorphism (-308G/A) of Tumor Necrosis Factor-Alpha (TNF-α) Gene and Asthma Risk: An Updated Meta-Analysis. Genet. Test. Mol. Biomark. 2019, 23, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.C.D.; Gomes, T.N.; Góis, I.A.F.; Oliveira, J.S.; Coelho, L.F.L.; Ferreira, G.P.; Silva, F.R.P.D.; Pereira, A.C.T.D.C. Association of single nucleotide polymorphisms in TNF-α (-308G/A and -238G/A) to dengue: Case-control and meta-analysis study. Cytokine 2020, 134, 155183. [Google Scholar] [CrossRef] [PubMed]
- Ferdosian, F.; Dastgheib, S.A.; Hosseini-Jangjou, S.H.; Nafei, Z.; Lookzadeh, M.H.; Noorishadkam, M.; Mirjalili, S.R.; Neamatzadeh, H. Association of TNF-α rs1800629, CASP3 rs72689236 and FCGR2A rs1801274 Polymorphisms with Susceptibility to Kawasaki Disease: A Comprehensive Meta-Analysis. Fetal Pediatr. Pathol. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.Z.; Ezzat, D.A. Association of-308G/A Polymorphism and Serum Level of TNF-α with Bronchial asthma in Children. Egypt. J. Immunol. 2018, 25, 117–124. [Google Scholar] [PubMed]
- Thriveni, K.; Raju, A.; Ramaswamy, G.; Krishnamurthy, S. Impact of gene polymorphism of TNF-α rs 1800629 and TNF-β rs 909253 on plasma levels of South Indian breast cancer patients. Indian J. Cancer. 2018, 55, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Hurtado, I.A.; Puebla-Pérez, A.M.; Delgado-Saucedo, J.I.; Figuera, L.E.; Zúñiga-González, G.M.; Gomez-Mariscal, K.; Ronquillo-Carreón, C.A.; Gallegos-Arreola, M.P. Association between TNF-α-308G>A and -238G>A gene polymorphisms and TNF-α serum levels in Mexican colorectal cancer patients. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Culjak, M.; Perkovic, M.N.; Uzun, S.; Strac, D.S.; Erjavec, G.N.; Leko, M.B.; Simic, G.; Tudor, L.; Konjevod, M.; Kozumplik, O.; et al. The Association between TNF-alpha, IL-1 alpha and IL-10 with Alzheimer’s Disease. Curr. Alzheimer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Sharif, D.; Jaf, M.; Amin, D.M. Effect of TNF-α -308G/A (rs1800629) Promoter Polymorphism on the Serum Level of TNF-α Among Iraqi Patients with Generalized Vitiligo. Clin. Cosmet. Investig. Dermatol. 2020, 13, 825–835, PMCID: PMC7671505. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR9&keywords=ccr9 (accessed on 26 February 2021).
- Pathak, M.; Lal, G. The Regulatory Function of CCR9+ Dendritic Cells in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 536326, PMCID: PMC7566413. [Google Scholar] [CrossRef]
- Biswas, S.; Bryant, R.V.; Travis, S. Interfering with leukocyte trafficking in Crohn’s disease. Best Pract. Res. Clin. Gastroenterol. 2019, 38–39, 1016. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CXCR6&keywords=cxcr6 (accessed on 26 February 2021).
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 2019, 216, 2748–2762, PMCID: PMC6888981. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=XCR1&keywords=XCR1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=XCL1&keywords=xcl1 (accessed on 26 February 2021).
- Lei, Y.; Takahama, Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012, 14, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR2&keywords=ccr2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR5&keywords=ccr5 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR3&keywords=ccr3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=PEDS1-UBE2V1 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/610994?search=tmem189&highlight=tmem189 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/602995?search=ube2v1&highlight=ube2v1 (accessed on 26 February 2021).
- Mackelprang, R.D.; Bamshad, M.J.; Chong, J.X.; Hou, X.; Buckingham, K.J.; Shively, K.; deBruyn, G.; Mugo, N.R.; Mullins, J.I.; McElrath, M.J.; et al. Whole genome sequencing of extreme phenotypes identifies variants in CD101 and UBE2V1 associated with increased risk of sexually acquired HIV-1. PLoS Pathog. 2017, 13, e1006703, Erratum in 2019, 15, e1007588. PMCID: PMC5690691. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL10&keywords=il10 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL17A&keywords=il17a (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL17F&keywords=il17f (accessed on 26 February 2021).
- Karcıoğlu Batur, L.; Hekim, N. Correlations of IL-6, IL-6R, IL-10 and IL-17 gene polymorphisms with the prevalence of COVID-2019 infection and its mortality rate. medRxiv 2020. Available online: https://www.researchsquare.com/article/rs-82662/v1 (accessed on 26 February 2021). [CrossRef]
- Gonçalves de Albuquerque, S.D.C.; da Costa Oliveira, C.N.; Vaitkevicius-Antão, V.; Silva, A.C.; Luna, C.F.; de Lorena, V.M.B.; de Paiva-Cavalcanti, M. Study of association of the rs2275913 IL-17A single nucleotide polymorphism and susceptibility to cutaneous leishmaniasis caused by Leishmania braziliensis. Cytokine 2019, 123, 154784. [Google Scholar] [CrossRef] [PubMed]
- Rolandelli, A.; Hernández Del Pino, R.E.; Pellegrini, J.M.; Tateosian, N.L.; Amiano, N.O.; de la Barrera, S.; Casco, N.; Gutiérrez, M.; Palmero, D.J.; García, V.E. The IL-17A rs2275913 single nucleotide polymorphism is associated with protection to tuberculosis but related to higher disease severity in Argentina. Sci. Rep. 2017, 7, 40666, PMCID: PMC5241634. [Google Scholar] [CrossRef]
- Yu, Z.G.; Wang, B.Z.; Li, J.; Ding, Z.L.; Wang, K. Association between interleukin-17 genetic polymorphisms and tuberculosis susceptibility: An updated meta-analysis. Int. J. Tuberc. Lung Dis. 2017, 21, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Cheng, B.; Ding, Y.; Wang, C.; Chen, J. Correlations of IL-17 and NF-κB gene polymorphisms with susceptibility and prognosis in acute respiratory distress syndrome in a chinese population. Biosci. Rep. 2019, 39, BSR20181987, PMCID: PMC6367126. [Google Scholar] [CrossRef]
- Zhai, C.; Li, S.; Feng, W.; Shi, W.; Wang, J.; Wang, Q.; Chai, L.; Zhang, Q.; Yan, X.; Li, M. Association of interleukin-17a rs2275913 gene polymorphism and asthma risk: A meta-analysis. Arch. Med. Sci. 2018, 14, 1204–1211, PMCID: PMC6209699. [Google Scholar] [CrossRef]
- Holster, A.; Teräsjärvi, J.; Lauhkonen, E.; Törmänen, S.; Helminen, M.; Koponen, P.; Korppi, M.; Peltola, V.; He, Q.; Nuolivirta, K. IL-17A gene polymorphism rs2275913 is associated with the development of asthma after bronchiolitis in infancy. Allergol. Int. 2018, 67, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Han, J.C.; Zhang, Y.J.; Qi, G.B.; Li, H.B.; Zhang, Y.J.; Cai, S. Single-Nucleotide Polymorphisms of IL-17 Gene Are Associated with Asthma Susceptibility in an Asian Population. Med. Sci. Monit. 2016, 22, 780–787, PMCID: PMC4793684. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=WSB1&keywords=wsb1v (accessed on 26 February 2021).
- Hu, J.; Li, C.; Wang, S.; Li, T.; Zhang, H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK Biobank data. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.11.05.20226761v1 (accessed on 26 February 2021). [CrossRef]
- Haque, M.; Kendal, J.K.; MacIsaac, R.M.; Demetrick, D.J. WSB1: From homeostasis to hypoxia. J. Biomed. Sci. 2016, 23, 61, PMCID: PMC4992216. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL21R&keywords=il-21 (accessed on 26 February 2021).
- Nara, H.; Onoda, T.; Rahman, M.; Araki, A.; Juliana, F.M.; Tanaka, N.; Asao, H. WSB-1, a novel IL-21 receptor binding molecule, enhances the maturation of IL-21 receptor. Cell Immunol. 2011, 269, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Ma, C.S. Regulation of the germinal center and humoral immunity by interleukin-21. J. Exp. Med. 2020, 217, e20191638, PMCID: PMC7037251. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.M.; Lukacher, A.E. IL-21 in Homeostasis of Resident Memory and Exhausted CD8 T Cells during Persistent Infection. Int. J. Mol. Sci. 2020, 21, 6966, PMCID: PMC7554897. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/608258 (accessed on 26 February 2021).
- Waumans, Y.; Baerts, L.; Kehoe, K.; Lambeir, A.M.; De Meester, I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front. Immunol. 2015, 6, 387, PMCID: PMC4528296. [Google Scholar] [CrossRef]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=DPP9&keywords=dpp9 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=DPP7&keywords=dpp7 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/610537?search=dpp7&highlight=dpp7 (accessed on 26 February 2021).
- Mele, D.A.; Bista, P.; Baez, D.V.; Huber, B.T. Dipeptidyl peptidase 2 is an essential survival factor in the regulation of cell quiescence. Cell Cycle 2009, 8, 2425–2434. [Google Scholar] [CrossRef] [PubMed]
- HIPC-CHI Signatures Project Team; HIPC-I Consortium. Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses. Sci Immunol. 2017, 2, eaal4656, PMCID: PMC5800877. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/602581?search=golga3&highlight=golga3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GOLGA3&keywords=golga3 (accessed on 26 February 2021).
- Ovsyannikova, I.G.; Kennedy, R.B.; O’Byrne, M.; Jacobson, R.M.; Pankratz, V.S.; Poland, G.A. Genome-wide association study of antibody response to smallpox vaccine. Vaccine 2012, 30, 4182–4189, PMCID: PMC3367131. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/609619?search=golga8b&highlight=golga8b (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GOLGA8B&keywords=golga8b (accessed on 26 February 2021).
- Available online: https://www.iaf.urmc.rochester.edu/results?search=rs200975425; https://www.iaf.urmc.rochester.edu/about.html (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/600168?search=mst1r&highlight=mst1r (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MST1R&keywords=MST1R (accessed on 26 February 2021).
- Chaudhuri, A. Regulation of Macrophage Polarization by RON Receptor Tyrosine Kinase Signaling. Front. Immunol. 2014, 5, 546, PMCID: PMC4215628. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.B.; Ray, M.; Lutz, M.; Sharda, D.; Xu, J.; Hankey, P.A. The RON receptor tyrosine kinase regulates IFN-gamma production and responses in innate immunity. J. Immunol. 2008, 181, 2303–2310. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.C.; Wilson, C.B.; Ray, M.; Correll, P.H. Macrophage-stimulating protein, the ligand for the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J. Immunol. 2004, 172, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Zhou, Y.Q.; Chen, Y.Q. Macrophage-stimulating protein and RON receptor tyrosine kinase: Potential regulators of macrophage inflammatory activities. Scand. J. Immunol. 2002, 56, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/613296?search=laptm4b&highlight=laptm4b (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=LAPTM4B&keywords=LAPTM4B (accessed on 26 February 2021).
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. LAPTMs regulate lisosomal function and interact with mucolipin 1: New clues for understanding mucolipidosis type IV. J. Cell Sci. 2011, 124, 459–468, PMCID: PMC3022000. [Google Scholar] [CrossRef][Green Version]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250, PMCID: PMC4455107. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/107741?search=apoe&highlight=apoe (accessed on 26 February 2021).
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188, PMCID: PMC2674716. [Google Scholar] [CrossRef]
- Kumar, N.; Janmohamed, K.; Nyhan, K.; Martins, S.S.; Cerda, M.; Hasin, D.; Scott, J.; Pates, R.; Ghandour, L.; Wazaify, M.; et al. Substance use and substance use disorder, in relation to COVID-19: Protocol for a scoping review. Syst. Rev. 2021, 10, 48, PMCID: PMC7857102. [Google Scholar] [CrossRef] [PubMed]
- Brickhill-Atkinson, M.; Hauck, F.R. Impact of COVID-19 on Resettled Refugees. Prim. Care 2021, 48, 57–66, PMCID: PMC7538065. [Google Scholar] [CrossRef]
- Fronteira, I.; Sidat, M.; Magalhães, J.P.; de Barros, F.P.C.; Delgado, A.P.; Correia, T.; Daniel-Ribeiro, C.T.; Ferrinho, P. The SARS-CoV-2 pandemic: A syndemic perspective. One Health 2021, 12, 100228, PMCID: PMC7887445. [Google Scholar] [CrossRef]
- Emeny, R.T.; Carpenter, D.O.; Lawrence, D.A. Health disparities: Intracellular consequences of social determinants of health. Toxicol. Appl. Pharmacol. 2021, 115444. [Google Scholar] [CrossRef] [PubMed]
- Bourdrel, T.; Annesi-Maesano, I.; Alahmad, B.; Maesano, C.N.; Bind, M.A. The impact of outdoor air pollution on COVID-19: A review of evidence from in vitro, animal, and human studies. Eur. Respir. Rev. 2021, 30, 200242, PMCID: PMC7879496. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, E.D.; Engin, A. The effect of environmental pollution on immune evasion checkpoints of SARS-CoV-2. Environ. Toxicol. Pharmacol. 2021, 81, 103520, PMCID: PMC7580701. [Google Scholar] [CrossRef]
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat. Immunol. 2020, 21, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Quinete, N.; Hauser-Davis, R.A. Drinking water pollutants may affect the immune system: Concerns regarding COVID-19 health effects. Environ. Sci. Pollut. Res. Int. 2021, 28, 1235–1246, PMCID: PMC7644792. [Google Scholar] [CrossRef] [PubMed]
- Zahra, A.; Sisu, C.; Silva, E.; De Aguiar Greca, S.C.; Randeva, H.S.; Chatha, K.; Kyrou, I.; Karteris, E. Is There a Link between Bisphenol A (BPA), a Key Endocrine Disruptor, and the Risk for SARS-CoV-2 Infection and Severe COVID-19? J. Clin. Med. 2020, 9, 3296, PMCID: PMC7602132. [Google Scholar] [CrossRef]
- Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; Graham, B.S.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. BioRxiv 2021. PMCID: PMC7852271. [Google Scholar] [CrossRef]
- Ortuso, F.; Mercatelli, D.; Guzzi, P.H.; Giorgi, F.M. Structural genetics of circulating variants affecting the SARS-CoV-2 spike/human ACE2 complex. J. Biomol. Struct. Dyn. 2021, 1–11, PMCID: PMC7885719. [Google Scholar] [CrossRef] [PubMed]
- Brookman, S.; Cook, J.; Zucherman, M.; Broughton, S.; Harman, K.; Gupta, A. Effect of the new SARS-CoV-2 variant B.1.1.7 on children and young people. Lancet Child. Adolesc. Health 2021. [Google Scholar] [CrossRef]
- Available online: https://covid19.who.int/ (accessed on 2 February 2021).
- Abduljalil, J.M.; Abduljalil, B.M. Epidemiology, genome, and clinical features of the pandemic SARS-CoV-2: A recent view. New Microbes New Infect. 2020, 35, 100672, PMCID: PMC7171182. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Lawrence, D.A. Susceptibility to COVID-19 in populations with health disparities: Posited involvement of mitochondrial disorder, socioeconomic stress, and pollutants. J. Biochem. Mol. Toxicol. 2020, e22626. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Harrison, E.M.; Ho, A.; Docherty, A.B.; Knight, S.R.; van Smeden, M.; Abubakar, I.; Lipman, M.; Quartagno, M.; Pius, R.; et al. Development and validation of the ISARIC 4C Deterioration model for adults hospitalised with COVID-19: A prospective cohort study. Lancet Respir. Med. 2021. PMCID: PMC7832571. [Google Scholar] [CrossRef] [PubMed]
- Márquez, E.J.; Chung, C.-H.; Marches, R.; Rossi, R.J.; Nehar-Belaid, D.; Eroglu, A.; Mellert, D.J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. Sexual-dimorphism in human immune system aging. Nat. Commun. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- Márquez, E.J.; Trowbridge, J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. The lethal sex gap: COVID-19. Immun. Ageing 2020, 17, 13, PMCID: PMC7240166. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Gadi, N.; Wu, S.C.; Spihlman, A.P.; Moulton, V.R. What’s Sex Got to Do With COVID-19? Gender-Based Differences in the Host Immune Response to Coronaviruses. Front. Immunol. 2020, 11, 2147, PMCID: PMC7485092. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/300386 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/300292?search=foxp3&highlight=foxp3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CXCR3&keywords=cxcr3 (accessed on 26 February 2021).
- Dattilo, M. The role of host defences in Covid 19 and treatments thereof. Mol. Med. 2020, 26, 90, PMCID: PMC7522454. [Google Scholar] [CrossRef]
- Van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 2020, 324, 1–11, PMCID: PMC7382021. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/147640?search=IFNB1&highlight=ifnb1 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/147571?search=isg15&highlight=isg15 (accessed on 26 February 2021).
- Aiello, A.; Accardi, G.; Candore, G.; Caruso, C.; Colomba, C.; Di Bona, D.; Duro, G.; Gambino, C.M.; Ligotti, M.E.; Pandey, J.P. Role of Immunogenetics in the Outcome of HCMV Infection: Implications for Ageing. Int. J. Mol. Sci. 2019, 20, 685, PMCID: PMC6386818. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Candore, G.; Accardi, G.; Caruso, C.; Colomba, C.; Duro, G.; Gambino, C.M.; Ligotti, M.E.; Di Bona, D. Translation of Basic Research into Clinics: Killer Immunoglobulin-like Receptors Genes in Autoimmune and Infectious Diseases. Curr. Pharm. Des. 2018, 24, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Pandey, J.P.; Puca, A.A. Genetics of exceptional longevity: Possible role of GM allotypes. Immun. Ageing 2018, 15, 25, PMCID: PMC6219196. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Netea, M.G. Trained Innate Immunity, Epigenetics, and Covid-19. N. Engl. J. Med. 2020, 383, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Papp, N.; Chou, J.T.; Hana, D.; Mackiewicz, A.; Kaczmarek, M. SARS-CoV-2: Pathogenesis, and Advancements in Diagnostics and Treatment. Front. Immunol. 2020, 11, 570927, PMCID: PMC7573101. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Luxami, V.; Tandon, R.; Paul, K. Recent Approaches of Repositioning and Traditional Drugs for the Treatment of COVID-19 Pandemic Outbreak. Mini Rev. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Jethava, Y.; Vikas, P. Repurposing Anti-Cancer Drugs for COVID-19 Treatment. Drug Des. Dev. Ther. 2020, 14, 5045–5058, PMCID: PMC7680713. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharm. Rep. 2020, 1–30, PMCID: PMC7474498. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Cordoba, V. Drug Repositioning for COVID-19. Colomb. Med. 2020, 51, e4279, PMCID: PMC7518729. [Google Scholar] [CrossRef]


{kind=link}
| Group/Locus | Variant | Outcome | p | OR (95% CI) | RR (95%CI) | Refs. |
|---|---|---|---|---|---|---|
| A | ||||||
| Susceptibility | 0.027 | 1.21 (1.02–1.43) | [21] | |||
| Susceptibility | * 0.04 | * 1.33 (1.02–1.73) | [24] | |||
| Susceptibility | 0.0024 | 1.23 (1.08–1.41) | [25] | |||
| Susceptibility | <0.001 | 1.09 (1.02–1.13) | [27] | |||
| Susceptibility (1) | <0.001 | 1.249 (1.114–1.440) | [26] | |||
| Susceptibility (2) | 0.03 | 1.3 (1.02–1.66) | [29] | |||
| Respiratory failure | * 1.48×10−4 | * 1.45 (1.20–1.75) | [28] | |||
| Mortality | 0.008 | 1.482 (1.113–1.972) | [21] | |||
| AB | ||||||
| Susceptibility | * 0.035 | * 1.37 (1.02–1.83) | [30] | |||
| B | ||||||
| Susceptibility | * 0.004 | * 1.28 (1.08–1.52) | [30] | |||
| 0 | ||||||
| Susceptibility | <0.001 | 0.67 (0.60–0.75) | [21] | |||
| Susceptibility | 0.0006 | 0.787 (0.69–0.90) | [25] | |||
| Susceptibility | <0.001 | 0.87 (0.82–0.91) | [27] | |||
| Susceptibility | * 0.007 | 0.84 (0.75–0.95) | [30] | |||
| Susceptibility (1) | <0.001 | 0.699 (0.635–0.770) | [26] | |||
| Respiratory failure | * 1.06×10−5 | * 0.65 (0.53–0.79) | [28] | |||
| Mortality | 0.014 | 0.660 (0.479–0.911) | [21] | |||
| Locus AB0 | ||||||
| rs657152 (A) | Respiratory failure | * 5.35×10−7 | * 1.39 (1.22–1.59) | [28] |
| Variable | Allele/Serotype | p | OR (95%CI) | Regression Coefficient | Growth Rate (95%CI) | Refs. |
|---|---|---|---|---|---|---|
| Incidence | B*44 | 0.05 | 0.1484 | 1.16 (1–1.35%) | [39] | |
| C*01 | 0.042 | 0.1747 | 1.19 (1.01–1.41%) | [39] | ||
| Susceptibility | B*15:27 | 0.030 | 3.59 (1.72–7.50) § | [45] | ||
| B46 | n.s. | [46] | ||||
| B22 | 0.032 | 1.71 (1.23–2.38) § | [46] | |||
| C*07:29 | 0.025 | 130.20 (5.28–3211) § | [45] | |||
| DQB1*06 | 0.0468 # | 1.96 (1.19–3.22) #§ | [47] | |||
| DRB1*08 | 0.010 # | 1.814 (1.151–2.860) # | [29] | |||
| Severity | A | n.s. | [28] | |||
| A*11:01 | 0.008 | 2.33 | [10] | |||
| B22 | n.s. | [46] | ||||
| B27 | n.s. | [46,48] | ||||
| B*27:07 | * 0.004 | [41] | ||||
| B46 | n.s. | [46] | ||||
| B*51:01 | 0.007 | 3.38 | [10] | |||
| C | n.s. | [28] | ||||
| C*14:02 | 0.003 | 4.75 | [10] | |||
| DPB1*03:01 | 0.037 | 0.09 | [10] | |||
| DQA1*01:01 | 0.039 | 6.05 | [10] | |||
| DQB1 | n.s. | [28] | ||||
| DQB1*06:02 | 0.016 | [41] | ||||
| DRB1 | n.s. | [28] | ||||
| DRB1*01:01 | 0.02 | 13.7 | [10] | |||
| DRB1*12:01 | 0.045 | 0.18 | [10] | |||
| DRB1*14:04 | 0.01 | 15.1 | [10] | |||
| DRB1*15:01 | 0.048 | [37] | ||||
| A*11 | 0.04 (1) | 7.693 (1.063–55.65) (1) | [40] | |||
| 0.02 (2) | 11.858 (1.524–92.273) (2) | [40] | ||||
| Mortality | C*01 | 0.04 (1) | 11.182 (1.053–118.7) (1) | [40] | ||
| 0.02 (2) | 17.604 (1.629–190.211) (2) | [40] | ||||
| DQB1*04 | 0.03 (1) | 9.963 (1.235–80.358) (1) | [40] | |||
| DRB1*08 | 0.01 # | 8.6 (1.7–43.9) # | [29] |
| Gene/Locus | Variant/ Position | Reference/ Other Allele | Altered/ Risk Allele | Protein Variant | Type | p | OR (95%CI) | Outcome | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| TLR3 | § 187003852 | AT | A | p.Ser339fs | pLOF | 0.01 | 8.28 (1.04–65.64) | severity | [113] |
| TLR3 | § 187005146 | G | A | p.Trp769 * | pLOF | ||||
| UNC93B1 | § 67770598 | C | A | p.Glu96 * | pLOF | ||||
| TBK1 | § 64875731 | C | T | p.Arg308 * | pLOF | ||||
| IRF7 | § 615095 | A | C | p.Arg7fs | pLOF | ||||
| IRF7 | § 614300 | G | A | p.Gln185 * | pLOF | ||||
| IRF7 | § 613966 | CGGGCTGGGGCCCG | C | p.Pro246fs | pLOF | ||||
| IRF7 | § 613353 | G | GC | p.Pro364fs | pLOF | ||||
| IFNAR2 | § 34621038 | AGATTGTTGGTTTT | A | p.Glu140fs | pLOF | ||||
| IFNAR2 | rs2236757 | G | A | 4.99 × 10−8 | 1.28 | severity | [117] | ||
| OAS3 | rs10735079 | G | A | 1.65 × 10−8 | 1.29 | severity | [117] | ||
| IFITM3 | rs12252 | T | C | # 0.0093 | # 6.37 | severity | [118] | ||
| 0.025 | 1.93 (1.09–3.46) | severity | [119] | ||||||
| PRKRA | I226N | 0.02 | severity | [120] | |||||
| TNF-α | rs1800629 | G | A | ° <0.001 | age > 60 | [121] | |||
| ° <0.001 | lymphopenia | [121] | |||||||
| ° 0.009 | high CRP | [121] | |||||||
| ° <0.001 | high ferritin | [121] | |||||||
| ° <0.001 | severity | [121] | |||||||
| † 0.045 | severity | [121] | |||||||
| 3p21.31 | rs11385942 | G | GA | 1.15 × 10−10 | 1.77 (1.48–2.11) | respiratory failure | [28] | ||
| 0.003 | 1.56 (1.17–2.01) | mechanical ventilation | [28] | ||||||
| TMEM189UBE2V1 | rs6020298 | G | A | 4.1 × 10–6 | 1.2 | severity | [10] | ||
| DPP9 | rs2109069 | G | A | 3.98 × 10−12 | 1.36 | severity | [117] | ||
| GOLGA8B | rs200975425 | C | T | 9.4 × 10–10 | 5.4 | susceptibility | [10] | ||
| LAPTM4B | P219L | 0.029 | [120] | ||||||
| P220L | |||||||||
| I109F | |||||||||
| P50T | |||||||||
| ApoE | e3 | e4 | 1.19 × 10–6 | 2.31 (1.65–3.24) | severity | [122] | |||
| ^ 0.009 | ^ 1.20 (1.05–1.37) | severity | [123] | ||||||
| 3.24 × 10−9 | 2.24 (1.72–2.93) | severity | [123] | ||||||
| 1.22 × 10–6 | 4.29 (2.38–7.72) | mortality | [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pojero, F.; Candore, G.; Caruso, C.; Di Bona, D.; Groneberg, D.A.; Ligotti, M.E.; Accardi, G.; Aiello, A. The Role of Immunogenetics in COVID-19. Int. J. Mol. Sci. 2021, 22, 2636. https://doi.org/10.3390/ijms22052636
Pojero F, Candore G, Caruso C, Di Bona D, Groneberg DA, Ligotti ME, Accardi G, Aiello A. The Role of Immunogenetics in COVID-19. International Journal of Molecular Sciences. 2021; 22(5):2636. https://doi.org/10.3390/ijms22052636
Chicago/Turabian StylePojero, Fanny, Giuseppina Candore, Calogero Caruso, Danilo Di Bona, David A. Groneberg, Mattia E. Ligotti, Giulia Accardi, and Anna Aiello. 2021. "The Role of Immunogenetics in COVID-19" International Journal of Molecular Sciences 22, no. 5: 2636. https://doi.org/10.3390/ijms22052636
APA StylePojero, F., Candore, G., Caruso, C., Di Bona, D., Groneberg, D. A., Ligotti, M. E., Accardi, G., & Aiello, A. (2021). The Role of Immunogenetics in COVID-19. International Journal of Molecular Sciences, 22(5), 2636. https://doi.org/10.3390/ijms22052636