
Two Siblings Homozygous for F508del-CFTR Have Varied Disease Phenotypes and Protein Biomarkers

 ,
,
Abstract
1. Introduction
2. Results
2.1. Clinical Description
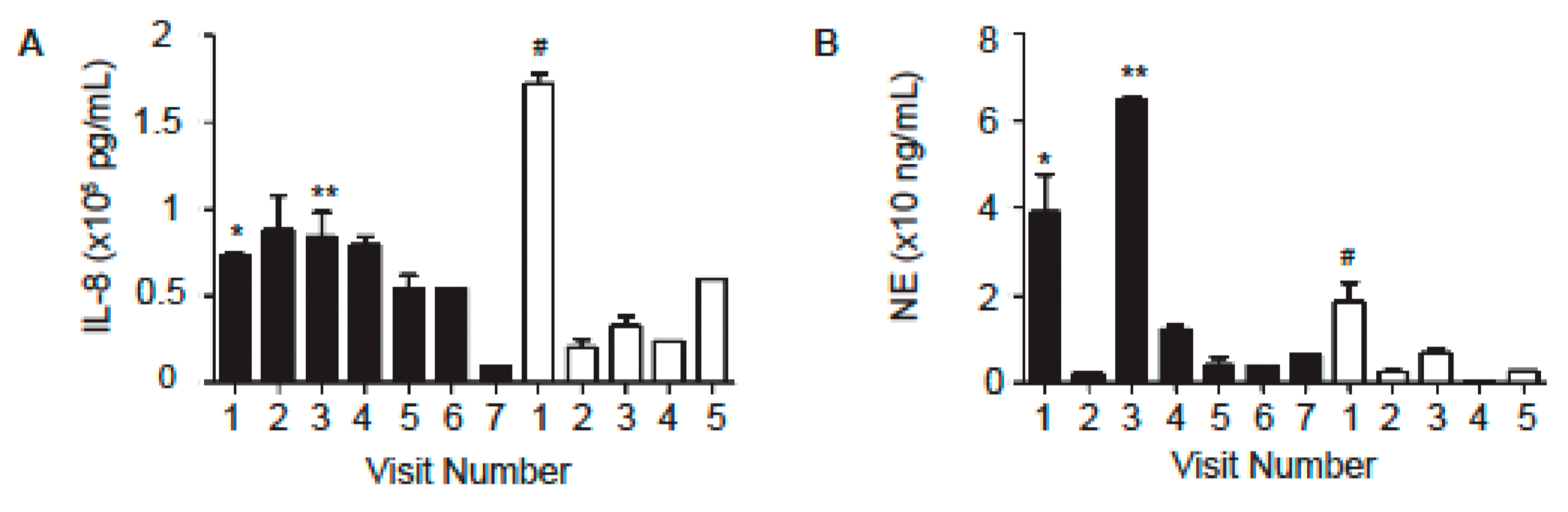
2.2. Levels of Neutrophil-Associated Markers in Sputum Samples
2.3. Levels of Other Inflammatory Mediators in Sputum Samples
3. Discussion
4. Materials and Methods
4.1. Subjects’ Characteristics
4.2. Measurements of IL-8 and NE Levels in Sputum Samples Using ELISA
4.3. ProcartaPlex™ Multiplex Immunoassay
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BAL fluid | broncho-alveolar lavage fluid |
| CF | cystic fibrosis |
| CFTR | CF transmembrane conductance regulator |
| DAMP | damage-associated molecular patterns |
| ELISA | enzyme-linked immunosorbent assay |
| ER | emergency room |
| FEV1 | forced expiratory volume in 1 s |
| FVC | forced vital capacity |
| GI | gastrointestinal |
| IL- | interleukin- |
| IP-10 | IFNγ-induced protein 10 kDa |
| MRSA | methicillin-resistant staphylococcus aureus |
| NE | neutrophilic elastase |
| PE | pulmonary exacerbation |
| PFT | pulmonary function test |
References
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation [US]. Available online: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/ (accessed on 30 July 2020).
- Rohlfs, E.M.; Zhou, Z.; Heim, R.A.; Nagan, N.; Rosenblum, L.S.; Flynn, K.; Scholl, T.; Akmaev, V.R.; Sirko-Osadsa, D.A.; Allitto, B.A.; et al. Cystic fibrosis carrier testing in an ethnically diverse US population. Clin. Chem. 2011, 57, 841–848. [Google Scholar] [CrossRef]
- Sugarman, E.A.; Rohlfs, E.M.; Silverman, L.M.; Allitto, B.A. CFTR mutation distribution among U.S. Hispanic and African American individuals: Evaluation in cystic fibrosis patient and carrier screening populations. Genet. Med. 2004, 6, 392–399. [Google Scholar] [CrossRef]
- Davies, J.C.; Alton, E.W.; Bush, A. Cystic fibrosis. BMJ 2007, 335, 1255–1259. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Banks-Schlegel, S.; Accurso, F.J.; Boucher, R.C.; Cutting, G.R.; Engelhardt, J.F.; Guggino, W.B.; Karp, C.L.; Knowles, M.R.; Kolls, J.K.; et al. Future directions in early cystic fibrosis lung disease research: An NHLBI workshop report. Am. J. Respir. Crit. Care Med. 2012, 185, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database. Available online: http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html (accessed on 10 December 2020).
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef]
- Corvol, H.; Blackman, S.M.; Boëlle, P.Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association me-ta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Schechter, M.S.; Shelton, B.J.; Margolis, P.A.; Fitzsimmons, S.C. The Association of Socioeconomic Status with Outcomes in Cystic Fibrosis Patients in the United States. Am. J. Respir. Crit. Care Med. 2001, 163, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.T.; Sarzynski, L.; Khalfoun, S.; Hayes, D., Jr.; Thompson, R.; Nicholson, L.; Long, F.; Castile, R.; Groner, J. Detrimental effects of secondhand smoke exposure on infants with cystic fibrosis. Pediatr. Pulmonol. 2015, 50, 25–34. [Google Scholar] [CrossRef]
- Blanchard, A.C.; Waters, V.J. Microbiology of Cystic Fibrosis Airway Disease. Semin. Respir. Crit. Care Med. 2019, 40, 727–736. [Google Scholar] [CrossRef]
- De Boeck, K.; Kent, L.; Davies, J.; Derichs, N.; Amaral, M.; Rowe, S.M. European Cystic Fibrosis Society Clinical Trial Network Standardisation Committee, CFTR biomarkers: Time for promotion to surrogate end-point. Eur. Respir. J. 2013, 41, 203–216. [Google Scholar] [CrossRef]
- Ramsey, K.A.; Schultz, A.; Stick, S.M. Biomarkers in Paediatric Cystic Fibrosis Lung Disease. Paediatr. Respir. Rev. 2015, 16, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hou, Y.; Sun, F.; Yang, Z.; Li, C. Dysregulated Chemokine Signaling in Cystic Fibrosis Lung Disease: A Potential Therapeutic Target. Curr. Drug Targets 2016, 17, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Laguna, T.A.; Williams, C.B.; Nunez, M.G.; Welchlin-Bradford, C.; Moen, C.E.; Reilly, C.S.; Wendt, C.H. Biomarkers of inflammation in infants with cystic fibrosis. Respir. Res. 2018, 19, 1–9. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between Pulmonary Function and Sputum Biomarkers in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung func-tion decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef]
- Kim, J.-S.; Okamoto, K.; Rubin, B.K. Pulmonary Function Is Negatively Correlated with Sputum Inflammatory Markers and Cough Clearability in Subjects with Cystic Fibrosis But Not Those With Chronic Bronchitis. Chest 2006, 129, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.S.; Schneider, P.; Kalled, S.L.; Wang, L.; Lefevre, E.A.; Cachero, T.G.; Mackay, F.; Bixler, S.A.; Zafari, M.; Liu, Z.-Y.; et al. Baff Binds to the Tumor Necrosis Factor Receptor–Like Molecule B Cell Maturation Antigen and Is Important for Maintaining the Peripheral B Cell Population. J. Exp. Med. 2000, 192, 129–136. [Google Scholar] [CrossRef]
- Do, R.K.G.; Hatada, E.; Lee, H.; Tourigny, M.R.; Hilbert, D.; Chen-Kiang, S. Attenuation of Apoptosis Underlies B Lymphocyte Stimulator Enhancement of Humoral Immune Response. J. Exp. Med. 2000, 192, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.; Groom, J.; Cachero, T.G.; Qian, F.; Schneider, P.; Tschopp, J.; Browning, J.L.; Mackay, F. BAFF mediates survival of peripheral immature B lymphocytes. J. Exp. Med. 2000, 192, 1453–1466. [Google Scholar] [CrossRef]
- Sutherland, A.P.R.; Ng, L.G.; Fletcher, C.A.; Shum, B.; Newton, R.A.; Grey, S.T.; Rolph, M.S.; Mackay, F.; Mackay, C.R. BAFF Augments Certain Th1-Associated Inflammatory Responses. J. Immunol. 2005, 174, 5537–5544. [Google Scholar] [CrossRef]
- Zhou, X.; Xia, Z.; Lan, Q.; Wang, J.; Su, W.; Han, Y.P.; Fan, H.; Liu, Z.; Stohl, W.; Zheng, S.G. BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS ONE 2011, 6, e23629. [Google Scholar] [CrossRef] [PubMed]
- Seys, L.J.M.; Verhamme, F.M.; Schinwald, A.; Hammad, H.; Cunoosamy, D.M.; Bantsimba-Malanda, C.; Sabirsh, A.; McCall, E.; Flavell, L.; Herbst, R.; et al. Role of B Cell–Activating Factor in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 706–718. [Google Scholar] [CrossRef]
- Laguna, T.A.; Williams, C.B.; Brandy, K.R.; Welchlin-Bradford, C.; Moen, C.E.; Reilly, C.S.; Wendt, C.H. Sputum club cell protein concentration is associated with pulmonary exacerbation in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 334–340. [Google Scholar] [CrossRef]
- Solomon, G.M.; Frederick, C.; Zhang, S.; Gaggar, A.; Harris, T.; Woodworth, B.A.; Steele, C.; Rowe, S.M. IP-10 Is a Potential Biomarker of Cystic Fibrosis Acute Pulmonary Exacerbations. PLoS ONE 2013, 8, e72398. [Google Scholar] [CrossRef]
- Ren, C.L.; Morgan, W.J.; Konstan, M.W.; Schechter, M.S.; Wagener, J.S.; Fisher, K.A.; Regelmann, W.E.; For the Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Presence of methicillin resistant Staphylococcus aureus in respiratory cultures from cystic fibrosis patients is associated with lower lung function. Pediatr. Pulmonol. 2007, 42, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Dasenbrook, E.C.; Merlo, C.A.; Diener-West, M.; Lechtzin, N.; Boyle, M.P. Persistent Methicillin-resistant Staphylococcus aureus and Rate of FEV 1 Decline in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 814–821. [Google Scholar] [CrossRef]
- King, J.; Brunel, S.F.; Warris, A. Aspergillus infections in cystic fibrosis. J. Infect. 2016, 72, S50–S55. [Google Scholar] [CrossRef] [PubMed]
- Gangell, C.; Gard, S.; Douglas, T.; Park, J.; De Klerk, N.; Keil, T.; Brennan, S.; Ranganathan, S.; Robins-Browne, R.; Sly, P.D.; et al. Inflammatory Responses to Individual Microorganisms in the Lungs of Children with Cystic Fibrosis. Clin. Infect. Dis. 2011, 53, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Modifier genes in Mendelian disorders: The example of cystic fibrosis. Ann. N. Y. Acad. Sci. 2010, 1214, 57–69. [Google Scholar] [CrossRef]
- Guillot, L.; Beucher, J.; Tabary, O.; Le Rouzic, P.; Clement, A.; Corvol, H. Lung disease modifier genes in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Website, Drug Development Pipeline—Clinical Trials Tool. Available online: https://www.cff.org/trials/pipeline (accessed on 8 September 2020).
- Pavord, I.D.; Pizzichini, M.M.; Pizzichini, E.; Hargreave, F.E. The use of induced sputum to investigate airway inflammation. Thorax 1997, 52, 498–501. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Visit # | Subject A | Subject B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 * | 2 | 3 † | 4 | 5 | 6 | 7 | 1 ¶ | 2 | 3 | 4 | 5 | |
| FEV1 (% predicated) | 45 | 38 | ND ‡ | 43 | 34 | ND ‡,§ | 39 | 53 | 73 | 57 | 48 | 90 |
| FVC (% predicated) | 65 | 58 | ND ‡ | 66 | 51 | ND ‡,‖ | 61 | 64 | 87 | 69 | 64 | 97 |
| Visit # | 1 * | 2 | 3 † | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Major symptoms | Cough, low-grade fevers (99 °F). | Worsen symptoms, cough with green sputum. | Felt too sick to perform PFT. | Increased congestion, sore throat, and cough. | Productive cough, low-grade fevers. | Fever (102 °F), cough, diffuse crackles and expiratory wheeze. | Increased cough without fever. |
| Microbes in sputum cultures | 3+ normal flora, 2+ MRSA, several Aspergillus fumigatus. | 4+ MRSA, 4+ normal flora, 1+ Stenotrophomonas maltophilia, 2+ Aspergillus fumigatus. | No sputum culture sent. | No sputum culture sent. | 4+ normal flora, 2+ MRSA. | 2+ MRSA, 2+ normal flora, few Stenotrophomonas maltophilia, few Aspergillus fumigatus. | No sputum culture sent. |
| Visit # | 1 * | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Major symptoms | Increased productive cough, blackish-tinged sputum, low-grade fevers, and rhinorrhea. | Mild increase in cough, decreased air exchange and wheezing. | Increased cough, sputum production, congestion, and low-grade fevers. | Reported worsened cough and sputum production. Crackles heard in right base. | GI illness developed 3–4 days prior to visit. |
| Microbes in sputum cultures | 2+ normal flora, 1+ non-mucoid Pseudomonas aeruginosa, 3+ MRSA, few Stenotrophomonas maltophilia. | No sputum culture obtained. | 4+ normal flora, 2+ Candida albicans, several E. Coli, few MRSA. | 1+ MRSA, few non-mucoid Pseudomonas aeruginosa, rare Candida albicans, few Klebsiella pneumoniae, 2+ normal flora. | No sputum culture obtained |
| Subject A | Subject B | |||||
|---|---|---|---|---|---|---|
| Average (Visit 1–7) | Non-PE (Visit 2, 4–7) | Preceding PE (Visit 1, 3) | Average (Visit 1–5) | Non-PE (Visit 2–4) | During PE (Visit 1) | |
| IL-8 (pg/mL) | 62,942 | 56,773 | 78,364 | 61,823 | 34,200 | 172,314 |
| NE (ng/mL) | 18.9 | 5.7 | 51.9 | 6.1 | 3 | 18.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wang, J.; Zhang, Y.H.; Gardner, T.E.; Fitzpatrick, E.A.; Zhang, W. Two Siblings Homozygous for F508del-CFTR Have Varied Disease Phenotypes and Protein Biomarkers. Int. J. Mol. Sci. 2021, 22, 2631. https://doi.org/10.3390/ijms22052631
Zhang Z, Wang J, Zhang YH, Gardner TE, Fitzpatrick EA, Zhang W. Two Siblings Homozygous for F508del-CFTR Have Varied Disease Phenotypes and Protein Biomarkers. International Journal of Molecular Sciences. 2021; 22(5):2631. https://doi.org/10.3390/ijms22052631
Chicago/Turabian StyleZhang, Zhihong, Jin Wang, Yanhui H. Zhang, Tonia E. Gardner, Elizabeth A. Fitzpatrick, and Weiqiang Zhang. 2021. "Two Siblings Homozygous for F508del-CFTR Have Varied Disease Phenotypes and Protein Biomarkers" International Journal of Molecular Sciences 22, no. 5: 2631. https://doi.org/10.3390/ijms22052631
APA StyleZhang, Z., Wang, J., Zhang, Y. H., Gardner, T. E., Fitzpatrick, E. A., & Zhang, W. (2021). Two Siblings Homozygous for F508del-CFTR Have Varied Disease Phenotypes and Protein Biomarkers. International Journal of Molecular Sciences, 22(5), 2631. https://doi.org/10.3390/ijms22052631