
Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility


Abstract
1. Introduction
2. Genetic Effects on Reproductive Traits
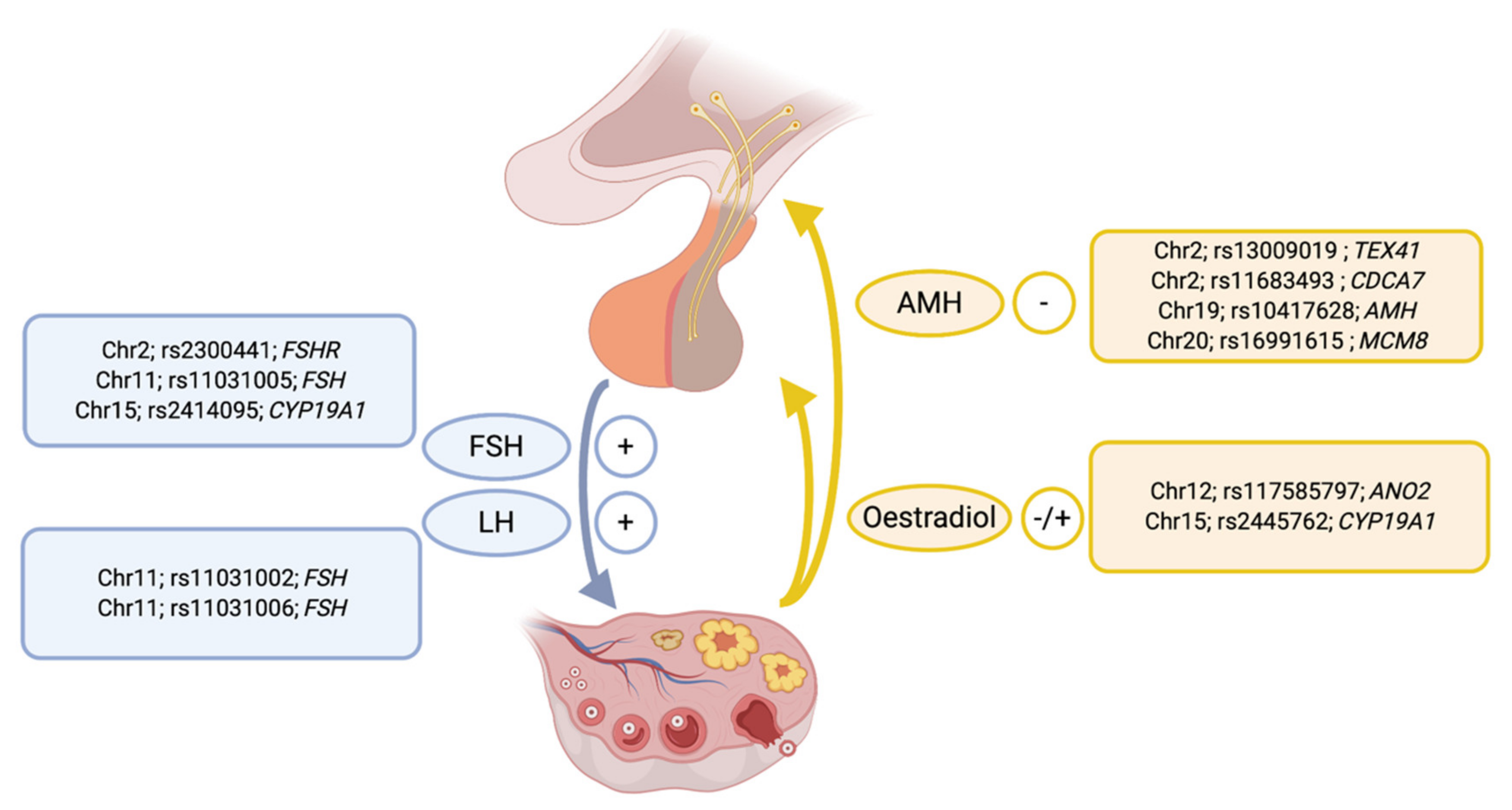
3. Genetic Variation Regulating Reproductive Hormone Concentrations
4. Shared Genetic Risk Factors between Reproductive Traits and Diseases
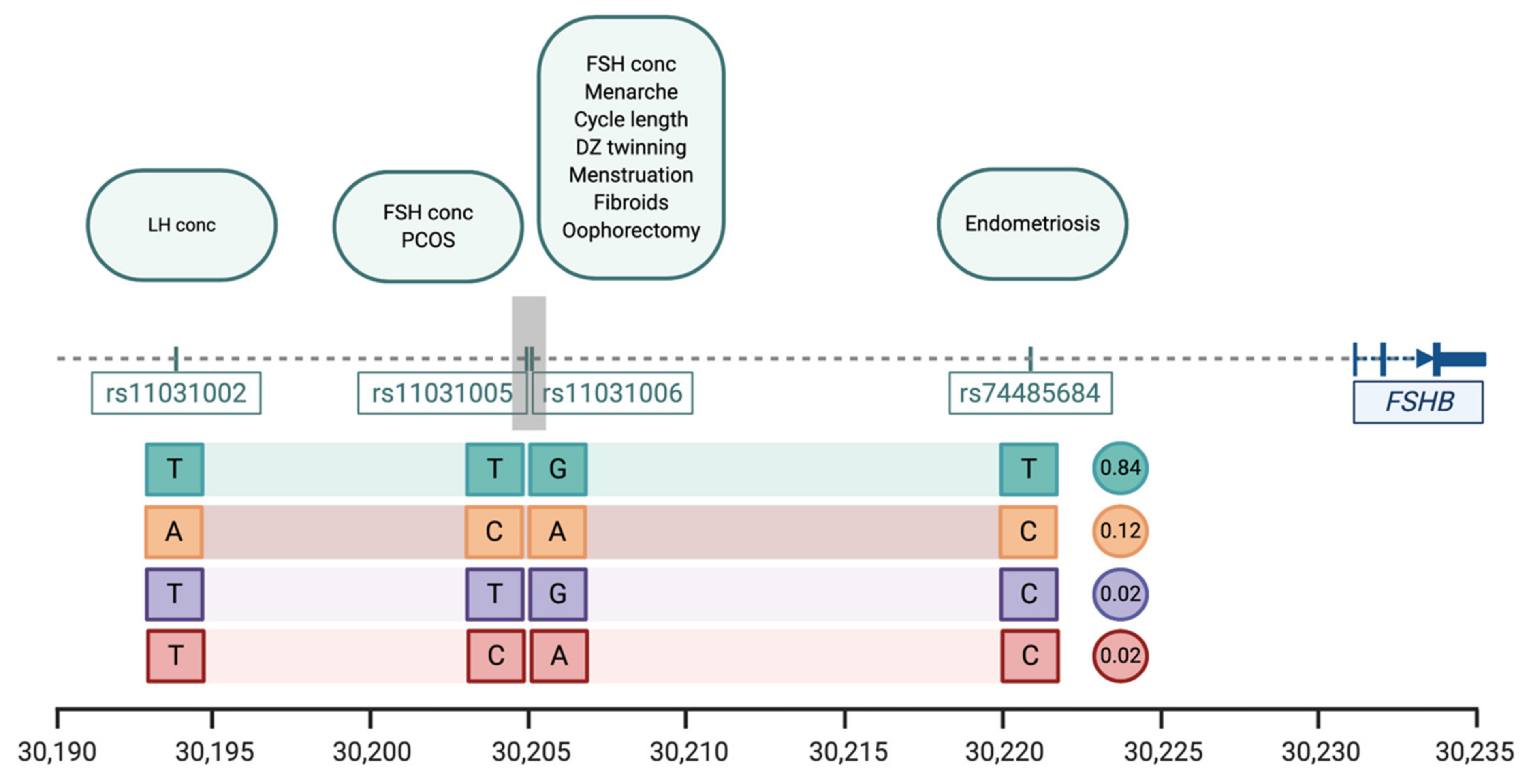
4.1. FSHB Locus on Chromosome 11
4.2. ESR1 Locus on Chromosome 6
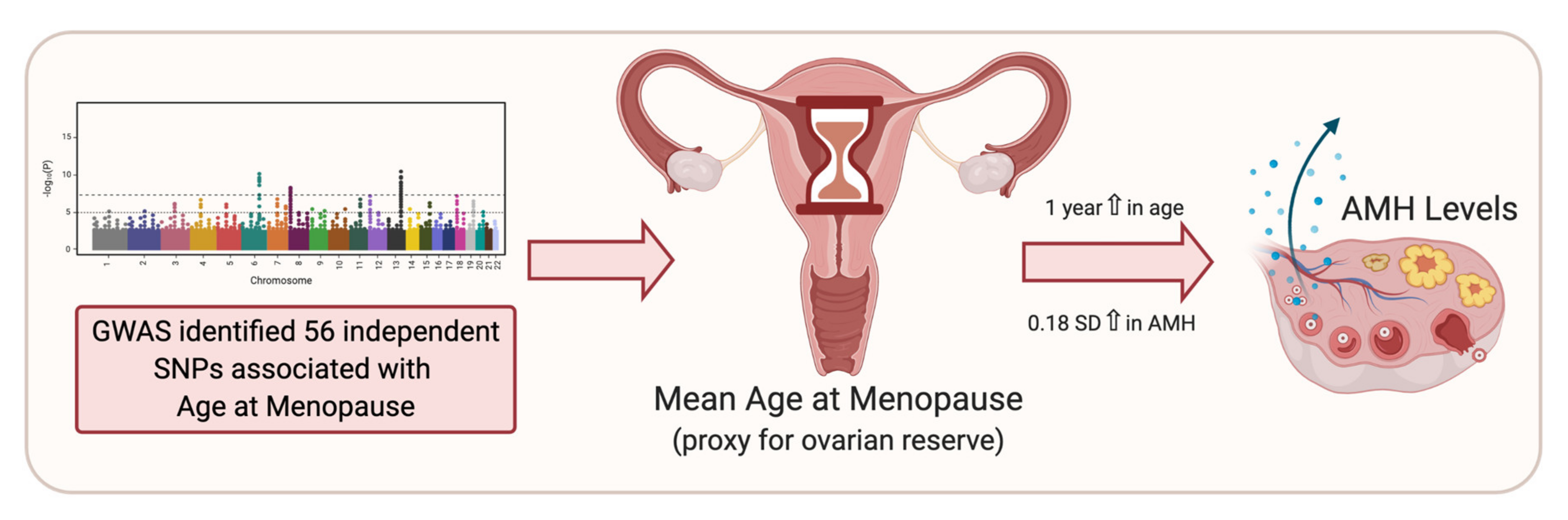
5. Age at Menopause
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fung, J.N.T.; Rogers, P.A.; Montgomery, G.W. Identifying the Biological Basis of GWAS Hits for Endometriosis1. Biol. Reprod. 2015, 92, 87. [Google Scholar] [CrossRef] [PubMed]
- Gajbhiye, R.; Fung, J.N.; Montgomery, G.W. Complex genetics of female fertility. npj Genom. Med. 2018, 3, 1–10. [Google Scholar] [CrossRef]
- Ruth, K.; Murray, A. Lessons from Genome-Wide Association Studies in Reproductive Medicine: Menopause. Semin. Reprod. Med. 2016, 34, 215–223. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Towne, B.; Czerwinski, S.A.; Demerath, E.W.; Blangero, J.; Roche, A.F.; Siervogel, R.M. Heritability of age at menarche in girls from the Fels Longitudinal Study. Am. J. Phys. Anthr. 2005, 128, 210–219. [Google Scholar] [CrossRef]
- Anderson, C.A.; Zhu, G.; Falchi, M.; Berg, S.M.V.D.; Treloar, S.A.; Spector, T.D.; Martin, N.G.; Boomsma, D.I.; Visscher, P.M.; Montgomery, G.W. A Genome-Wide Linkage Scan for Age at Menarche in Three Populations of European Descent. J. Clin. Endocrinol. Metab. 2008, 93, 3965–3970. [Google Scholar] [CrossRef]
- Morris, D.H.; Jones, M.E.; Schoemaker, M.J.; Ashworth, A.; Swerdlow, A.J. Familial concordance for age at menarche: Analyses from the Breakthrough Generations Study. Paediatr. Perinat. Epidemiol. 2011, 25, 306–311. [Google Scholar] [CrossRef]
- Kirk, K.M.; Blomberg, S.P.; Duffy, D.L.; Heath, A.C.; Owens, I.P.; Martin, N.G. Natural selection and quantitative genetics of life-history traits in Western women: A twin study. Evolution 2001, 55, 423–435. [Google Scholar] [CrossRef]
- Hur, Y.-M.; Jin, H.-J.; Lee, S. Heritability of Age at Menarche in South Korean Female Twins. Twin Res. Hum. Genet. 2019, 22, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Van Asselt, K.M.; Kok, H.S.; Pearson, P.L.; Dubas, J.S.; Peeters, P.H.; Velde, E.R.T.; Van Noord, P.A. Heritability of menopausal age in mothers and daughters. Fertil. Steril. 2004, 82, 1348–1351. [Google Scholar] [CrossRef]
- Snieder, H.; MacGregor, A.J.; Spector, T.D. Genes Control the Cessation of a Woman’s Reproductive Life: A Twin Study of Hysterectomy and Age at Menopause1. J. Clin. Endocrinol. Metab. 1998, 83, 1875–1880. [Google Scholar] [CrossRef]
- Murabito, J.M.; Yang, Q.; Fox, C.; Wilson, P.W.F.; Cupples, L.A. Heritability of Age at Natural Menopause in the Framingham Heart Study. J. Clin. Endocrinol. Metab. 2005, 90, 3427–3430. [Google Scholar] [CrossRef]
- Saha, R.; Pettersson, H.J.; Svedberg, P.; Olovsson, M.; Bergqvist, A.; Marions, L.; Tornvall, P.; Kuja-Halkola, R. Heritability of endometriosis. Fertil. Steril. 2015, 104, 947–952. [Google Scholar] [CrossRef]
- Treloar, S.A.; O’Connor, D.T.; O’Connor, V.M.; Martin, N.G. Genetic influences on endometriosis in an Australian twin sample. Fertil. Steril. 1999, 71, 701–710. [Google Scholar] [CrossRef]
- Luoto, R.; Kaprio, J.; Rutanen, E.-M.; Taipale, P.; Perola, M.; Koskenvuo, M. Heritability and risk factors of uterine fibroids — The Finnish Twin Cohort Study. Maturitas 2000, 37, 15–26. [Google Scholar] [CrossRef]
- Vink, J.M.; Sadrzadeh, S.; Lambalk, C.B.; Boomsma, D.I. Heritability of Polycystic Ovary Syndrome in a Dutch Twin-Family Study. J. Clin. Endocrinol. Metab. 2006, 91, 2100–2104. [Google Scholar] [CrossRef]
- Svejme, O.; Ahlborg, H.; Nilsson, J.-Å; Karlsson, M. Early menopause and risk of osteoporosis, fracture and mortality: A 34-year prospective observational study in 390 women. BJOG: Int. J. Obstet. Gynaecol. 2012, 119, 810–816. [Google Scholar] [CrossRef]
- Salonen Ros, H.; Lichtenstein, P.; Lipworth, L.; Cnattingius, S. Genetic effects on the liability of developing pre-eclampsia and gestational hypertension. Am. J. Med. Genet. 2000, 91, 256–260. [Google Scholar] [CrossRef]
- Laisk, T.; Kukuskina, V.; Palmer, D.; Laber, S.; Chen, C.Y.; Ferreira, T.; Rahmioglu, N.; Zondervan, K.; Becker, C.; Smoller, J.W.; et al. Large scale meta-analysis highlights the hypothalamic-pituitary-gonadal (HPG) axis in the genetic regulation of menstrual cycle length. Hum. Mol. Genet. 2018, 27, 4323–4332. [Google Scholar] [PubMed]
- Elks, C.E.; Perry, J.R.; Sulem, P.; Chasman, D.I.; Franceschini, N.; He, C.; Lunetta, K.L.; Visser, J.A.; Byrne, E.M.; Cousminer, D.L.; et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 2010, 42, 1077–1085. [Google Scholar] [CrossRef]
- Perry, J.R.B.; Australian Ovarian Cancer Study; Day, F.R.; Elks, C.E.; Sulem, P.; Thompson, D.J.; Ferreira, T.; He, C.; Chasman, D.I.; Esko, T.; et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nat. Cell Biol. 2014, 514, 92–97. [Google Scholar] [CrossRef]
- Day, F.R.; The LifeLines Cohort Study; Thompson, D.J.; Helgason, H.; Chasman, D.I.; Finucane, H.; Sulem, P.; Ruth, K.S.; Whalen, S.; Sarkar, A.K.; et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Genet. 2017, 49, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Uno, S.; Zembutsu, H.; Hirasawa, A.; Takahashi, A.; Kubo, M.; Akahane, T.; Aoki, D.; Kamatani, N.; Hirata, K.; Nakamura, Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010, 42, 707–710. [Google Scholar] [CrossRef]
- Painter, J.N.; Anderson, C.A.; Nyholt, D.R.; Macgregor, S.; Lin, J.; Lee, S.H.; Lambert, A.; Zhao, Z.Z.; Roseman, F.; Guo, Q.; et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011, 43, 51–54. [Google Scholar] [CrossRef]
- Nyholt, D.R.; Low, S.-K.; Anderson, C.A.; Painter, J.N.; Uno, S.; Morris, A.P.; MacGregor, S.; Gordon, S.D.; Henders, A.K.; Martin, N.G.; et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat. Genet. 2012, 44, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; iPSYCH-SSI-Broad Group; Steinthorsdottir, V.; Morris, A.P.; Fassbender, A.; Rahmioglu, N.; De Vivo, I.; Buring, J.E.; Zhang, F.; Edwards, T.L.; et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun. 2017, 8, 15539. [Google Scholar] [CrossRef] [PubMed]
- Stolk, L.; Zhai, G.; Van Meurs, J.B.J.; Verbiest, M.M.P.J.; Visser, J.A.; Estrada, K.; Rivadeneira, F.; Williams, F.M.; Cherkas, L.; Deloukas, P.; et al. Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nat. Genet. 2009, 41, 645–647. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Kraft, P.; Chen, C.; Buring, J.E.; Paré, G.; Hankinson, S.E.; Chanock, S.J.; Ridker, P.M.; Hunter, D.J.; Chasman, D.I. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat. Genet. 2009, 41, 724–728. [Google Scholar] [CrossRef]
- Stolk, L.; Perry, J.R.B.; Chasman, D.I.; He, C.; Mangino, M.; Sulem, P.; Barbalic, M.; Broer, L.; Byrne, E.M.; Ernst, F.; et al. Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat. Genet. 2012, 44, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Day, F.R.; Ruth, K.S.; Thompson, D.J.; Lunetta, K.L.; Pervjakova, N.; Chasman, D.I.; Stolk, L.; Finucane, H.K.; Sulem, P.; Bulik-Sullivan, B.; et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 2015, 47, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Tropf, F.C.; Stulp, G.; Barban, N.; Visscher, P.M.; Yang, J.; Snieder, H.; Mills, M.C. Human Fertility, Molecular Genetics, and Natural Selection in Modern Societies. PLoS ONE 2015, 10, e0126821. [Google Scholar] [CrossRef]
- Lee, S.H.; Harold, D.; Nyholt, D.R.; ANZGene Consortium; Goddard, M.E.; Zondervan, K.T.; Williams, J.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum. Mol. Genet. 2013, 22, 832–841. [Google Scholar] [CrossRef]
- Bray, M.J.; Davis, L.K.; Torstenson, E.S.; Jones, S.H.; Edwards, T.L.; Edwards, D.R.V. Estimating Uterine Fibroid SNP-Based Heritability in European American Women with Imaging-Confirmed Fibroids. Hum. Hered. 2019, 84, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Laisk, T.; Soares, A.L.G.; Ferreira, T.; Painter, J.N.; Censin, J.C.; Laber, S.; Bacelis, J.; Chen, C.-Y.; Lepamets, M.; Lin, K.; et al. The genetic architecture of sporadic and multiple consecutive miscarriage. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Hayes, M.G.; Reproductive Medicine Network; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.K.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef]
- Ruth, K.S.; Campbell, P.J.; Chew, S.; Lim, E.M.; Hadlow, N.C.; Stuckey, B.G.A.; Brown, S.J.; Feenstra, B.; Joseph, J.; Surdulescu, G.L.; et al. Genome-wide association study with 1000 genomes imputation identifies signals for nine sex hormone-related phenotypes. Eur. J. Hum. Genet. 2016, 24, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tian, Y.; Gao, X.; Cui, L.; Ning, Y.; Cao, Y.; Chen, Y.; Peng, F.; You, L.; Liu, F.; et al. A genome-wide association study identifies FSHR rs2300441 associated with follicle-stimulating hormone levels. Clin. Genet. 2020, 97, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tao, S.; Gao, Y.; Zhang, J.; Hu, Y.; Mo, L.; Kim, S.-T.; Yang, X.; Tan, A.; Zhang, H.; et al. Genome-wide association study of sex hormones, gonadotropins and sex hormone–binding protein in Chinese men. J. Med Genet. 2013, 50, 794–801. [Google Scholar] [CrossRef]
- Verdiesen, R.M.; van der Schouw, Y.T.; van Gils, C.H.; Verschuren, W.M.; Broekmans, F.J.; Borges, M.C.; Soares, A.L.; Lawlor, D.A.; Eliassen, A.H.; Kraft, P.; et al. Genome-wide association study meta-analysis identifies three novel loci for circulating anti-Mullerian hormone levels in women. medRxiv 2020. [Google Scholar] [CrossRef]
- Ruth, K.S.; Soares, A.L.G.; Borges, M.-C.; Eliassen, A.H.; Hankinson, S.E.; Jones, M.E.; Kraft, P.; Nichols, H.B.; Sandler, D.P.; Schoemaker, M.J.; et al. Genome-wide association study of anti-Müllerian hormone levels in pre-menopausal women of late reproductive age and relationship with genetic determinants of reproductive lifespan. Hum. Mol. Genet. 2019, 28, 1392–1401. [Google Scholar] [CrossRef]
- Burger, L.L.; Haisenleder, D.J.; Aylor, K.W.; Dalkin, A.C.; Prendergast, K.A.; Marshall, J.C. Regulation of luteinizing hormone-beta and follicle-stimulating hormone (FSH)-beta gene transcription by androgens: Testosterone directly stimulates FSH-beta transcription independent from its role on follistatin gene expression. Endocrinology 2004, 145, 71–78. [Google Scholar] [CrossRef][Green Version]
- Stamatiades, G.A.; Kaiser, U.B. Gonadotropin regulation by pulsatile GnRH: Signaling and gene expression. Mol. Cell. Endocrinol. 2018, 463, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Mbarek, H.; Steinberg, S.; Nyholt, D.R.; Gordon, S.D.; Miller, M.B.; McRae, A.F.; Hottenga, J.J.; Day, F.R.; Willemsen, G.; De Geus, E.J.; et al. Identification of Common Genetic Variants Influencing Spontaneous Dizygotic Twinning and Female Fertility. Am. J. Hum. Genet. 2016, 98, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Day, F.; Karaderi, T.; Jones, M.R.; Meun, C.; He, C.; Drong, A.; Kraft, P.; Lin, N.; Huang, H.; Broer, L.; et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet. 2018, 14, e1007813. [Google Scholar] [CrossRef]
- Trevisan, C.M.; De Oliveira, R.; Christofolini, D.M.; Barbosa, C.P.; Bianco, B. Effects of a Polymorphism in the Promoter Region of the Follicle-Stimulating Hormone Subunit Beta (FSHB) Gene on Female Reproductive Outcomes. Genet. Test. Mol. Biomarkers 2019, 23, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Grigorova, M.; Punab, M.; Ausmees, K.; Laan, M. FSHB promoter polymorphism within evolutionary conserved element is associated with serum FSH level in men. Hum. Reprod. 2008, 23, 2160–2166. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Bohaczuk, S.C.; Thackray, V.G.; Shen, J.; Skowronska-Krawczyk, D.; Mellon, P.L. FSHB Transcription is Regulated by a Novel 5′ Distal Enhancer With a Fertility-Associated Single Nucleotide Polymorphism. Endocrinol. 2021, 162, 181. [Google Scholar] [CrossRef]
- Jameson, J.L. Of Mice and Men: The Tale of Steroidogenic Factor-1. J. Clin. Endocrinol. Metab. 2004, 89, 5927–5929. [Google Scholar] [CrossRef][Green Version]
- Day, F.R.; Hinds, D.A.; Tung, J.Y.; Stolk, L.; Styrkarsdottir, U.; Saxena, R.; Bjonnes, A.; Broer, L.; Dunger, D.B.; Halldorsson, B.V.; et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat. Commun. 2015, 6, 8464. [Google Scholar] [CrossRef]
- Paul, P.; Hoorn, T.V.D.; Jongsma, M.L.; Bakker, M.J.; Hengeveld, R.; Janssen, L.; Cresswell, P.; Egan, D.A.; Van Ham, M.; Brinke, A.T.; et al. A Genome-wide Multidimensional RNAi Screen Reveals Pathways Controlling MHC Class II Antigen Presentation. Cell 2011, 145, 268–283. [Google Scholar] [CrossRef]
- GTEx Consortium. Erratum: Genetic effects on gene expression across human tissues. Nature 2018, 553, 530. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.S.; Mäkinen, N.; Harris, H.R.; Rahmioglu, N.; Uimari, O.; Cook, J.P.; Shigesi, N.; Ferreira, T.; Velez-Edwards, D.R.; Edwards, T.L.; et al. Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Enns, D.L.; Tiidus, P.M. The Influence of Estrogen on Skeletal Muscle. Sports Med. 2010, 40, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.F.; Cambiasso, M.J.; Holschbach, M.A.; Cabrera, R. Oestrogens and Progestagens: Synthesis and Action in the Brain. J. Neuroendocr. 2016, 28, 7. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef]
- Ponglikitmongkol, M.; Green, S.; Chambon, P. Genomic organization of the human oestrogen receptor gene. EMBO J. 1988, 7, 3385–3388. [Google Scholar] [CrossRef]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-A. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern1. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Gustafsson, J.A. Estrogen signaling: A subtle balance between ER alpha and ER beta. Mol. Interv. 2003, 3, 281–392. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Zheng, W.; Long, J.; Gao, Y.-T.; Li, C.; Zheng, Y.; Xiang, Y.-B.; Wen, W.; Levy, S.; Deming, S.L.; Haines, J.L.; et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 2009, 41, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Barban, N.; Jansen, R.; De Vlaming, R.; Vaez, A.; Mandemakers, J.J.; Tropf, F.C.; Shen, X.; Wilson, J.F.; Chasman, D.I.; BIOS Consortium; et al. Genome-wide analysis identifies 12 loci influencing human reproductive behavior. Nat. Genet. 2016, 48, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Bahia, W.; Soltani, I.; Haddad, A.; Soua, A.; Radhouani, A.; Mahdhi, A.; Ferchichi, S. Association of genetic variants in Estrogen receptor (ESR)1 and ESR2 with susceptibility to recurrent pregnancy loss in Tunisian women: A case control study. Gene 2020, 736, 144406. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, I.; Zois, C.; Ioannidis, J.P.A.; Tsatsoulis, A. Association of polymorphisms of the oestrogen receptor α gene with the age of menarche. Hum. Reprod. 2002, 17, 1101–1105. [Google Scholar] [CrossRef]
- Vagnini, L.D.; Renzi, A.; Petersen, B.; Canas, M.D.C.T.; Petersen, C.G.; Mauri, A.L.; Mattila, M.C.; Ricci, J.; Dieamant, F.; Oliveira, J.B.A.; et al. Association between estrogen receptor 1 (ESR1) and leukemia inhibitory factor (LIF) polymorphisms can help in the prediction of recurrent implantation failure. Fertil. Steril. 2019, 111, 527–534. [Google Scholar] [CrossRef]
- Wedrén, S.; Lovmar, L.; Humphreys, K.; Magnusson, C.; Melhus, H.; Syvänen, A.-C.; Kindmark, A.; Landegren, U.; Fermér, M.L.; Stiger, F.; et al. Estrogen receptor alpha gene polymorphism and endometrial cancer risk—A case-control study. BMC Cancer 2008, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Han, R.; Chen, M.; Yuan, Y.; Hu, X.; Ma, Y.; Wu, M.; Zhang, X.; Wang, M.; Jiang, S.; et al. Associations of Estrogen Receptor Alpha Gene Polymorphisms with Type 2 Diabetes Mellitus and Metabolic Syndrome: A Systematic Review and Meta-Analysis. Horm. Metab. Res. 2018, 50, 469–477. [Google Scholar] [CrossRef]
- Zhou, A.; Liu, X.; Xia, T.; Li, F.; Wang, J.; Li, J. Estrogen receptor alpha gene (ESR1) polymorphism and its interaction with smoking and drinking contribute to susceptibility of systemic lupus erythematosus. Immunol. Res. 2017, 65, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; O’Mara, T.A.; Thompson, D.J.; Painter, J.N.; Attia, J.; Black, A.; Brinton, L.; Chanock, S.; Chen, C.; Cheng, T.H.; et al. GWAS meta-analysis of 16 852 women identifies new susceptibility locus for endometrial cancer. Hum. Mol. Genet. 2016, 25, 2612–2620. [Google Scholar] [CrossRef]
- Cheng, T.H.; National Study of Endometrial Cancer Genetics Group (NSECG); Thompson, D.J.; O’Mara, T.A.; Painter, J.N.; Glubb, D.M.; Flach, S.; Lewis, A.; French, J.D.; Freeman-Mills, L.; et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat. Genet. 2016, 48, 667–674. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Bolla, M.K.; et al. Identification of nine new susceptibility loci for endometrial cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Xue, A.; eQTLGen Consortium; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Marla, S.; Mortlock, S.; Houshdaran, S.; Fung, J.; McKinnon, B.; Holdsworth-Carson, S.J.; Girling, J.E.; Rogers, P.A.W.; Giudice, L.C.; Montgomery, G.W. Genetic risk factors for endometriosis near estrogen receptor 1 and co-expression of genes in this region in endometrium. Mol. Hum. Reprod. 2021, 27, gaaa082. [Google Scholar] [CrossRef] [PubMed]
- Lisabeth Lynda, D.; Beiser Alexa, S.; Brown Devin, L.; Murabito Joanne, M.; Kelly-Hayes, M.; Wolf Philip, A. Age at Natural Menopause and Risk of Ischemic Stroke. Stroke 2009, 40, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Atsma, F.; Bartelink, M.-L.E.L.; Grobbee, D.E.; Van Der Schouw, Y.T. Postmenopausal status and early menopause as independent risk factors for cardiovascular disease: a meta-analysis. Menopause 2006, 13, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Mondul, A.M.; Rodriguez, C.; Jacobs, E.J.; Calle, E.E. Age at Natural Menopause and Cause-specific Mortality. Am. J. Epidemiol. 2005, 162, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-C.; Trichopoulos, D.; Katsouyanni, K.; Yuasa, S. Age at menarche, age at menopause, height and obesity as risk factors for breast cancer: Associations and interactions in an international case-control study. Int. J. Cancer 1990, 46, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; La Vecchia, C.; Booth, M.; Tzonou, A.; Negri, E.; Parazzini, F.; Trichopoulos, D.; Beral, V. Pooled analysis of 3 european case-control studies of ovarian cancer: II. Age at menarche and at menopause. Int. J. Cancer 1991, 49, 57–60. [Google Scholar] [CrossRef]
- Wentzensen, N.; Poole, E.M.; Trabert, B.; White, E.; Arslan, A.A.; Patel, A.V.; Setiawan, V.W.; Visvanathan, K.; Weiderpass, E.; Adami, H.-O.; et al. Ovarian Cancer Risk Factors by Histologic Subtype: An Analysis From the Ovarian Cancer Cohort Consortium. J. Clin. Oncol. 2016, 34, 2888–2898. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, W.; Liu, H.; Zhang, D. Age at Menopause and Risk of Developing Endometrial Cancer: A Meta-Analysis. BioMed Res. Int. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Sheehan, N.A.; Didelez, V.; Burton, P.R.; Tobin, M.D. Mendelian Randomisation and Causal Inference in Observational Epidemiology. PLoS Med. 2008, 5, e177. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lv, J.; Qiu, X.; An, Y. Integrating genome-wide association and eQTLs studies identifies the genes associated with age at menarche and age at natural menopause. PLoS ONE 2019, 14, e0213953. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.A.; Schipper, I.; Laven, J.S.E.; Themmen, A.P.N. Anti-Müllerian hormone: An ovarian reserve marker in primary ovarian insufficiency. Nat. Rev. Endocrinol. 2012, 8, 331–341. [Google Scholar] [CrossRef]
- McRae, A.F.; Powell, J.E.; Henders, A.K.; Bowdler, L.; Hemani, G.; Shah, S.; Painter, J.N.; Martin, N.G.; Visscher, P.M.; Montgomery, G.W. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014, 15, R73. [Google Scholar] [CrossRef]
- Mortlock, S.; Restuadi, R.; Levien, R.; Girling, J.E.; Holdsworth-Carson, S.J.; Healey, M.; Zhu, Z.; Qi, T.; Wu, Y.; Lukowski, S.W.; et al. Genetic regulation of methylation in human endometrium and blood and gene targets for reproductive diseases. Clin. Epigenetics 2019, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trait | Heritability Estimate (95% CI) * | Reference |
|---|---|---|
| Age at Menarche | 0.49 (0.24–0.73) | [6] |
| 0.72 | [7] | |
| 0.57 (0.53–0.61) | [8] | |
| 0.51 (0.46–0.55) | [9] | |
| 0.72 (0.67–0.76) | [10] | |
| Age at First Reproduction | 0.24 (0.08–0.43) | [9] |
| Age at Menopause | 0.45 (0.16–0.58) | [9] |
| 0.44 (0.36–0.50) | [11] | |
| 0.63 (0.53–0.71) | [12] | |
| 0.52 (0.35–0.69) | [13] | |
| Hysterectomy | 0.59 (0.43–0.72) | [12] |
| Endometriosis | 0.47 (0.36–0.57) | [14] |
| 0.51 (0.36–0.66) | [15] | |
| Uterine Fibroids | 0.55 (0.46–0.63) & | [16] |
| 0.69 (0.49–0.83) $ | [12] | |
| PCOS | 0.79 | [17] |
| Pre-eclampsia | 0.31 (0.09–0.45) | [18] |
| 0.54 (0.00–0.71) | [19] | |
| Recurrent pregnancy loss | 0.29 (0.20–0.38) | [20] |
| Trait | SNP Heritability Estimate (Standard Error) | Reference |
|---|---|---|
| Age at menarche | 0.32 (0.01) | [23] |
| Age at first reproduction | 0.15 (0.04) | [32] |
| Age of menopause | 0.06 (0.02) | [31] |
| Endometriosis | 0.26 (0.04) | [33] |
| Uterine fibroids | 0.33 (0.18) | [34] |
| Recurrent pregnancy loss | 0.02 (0.40) | [35] |
| Trait | SNP | Position * | Pval | Effect Alleles & | Study |
|---|---|---|---|---|---|
| Menstrual cycle length | rs11031006 | 30,204,981 | 1.1 × 10−38 | A > G | [20] |
| Age at menarche | rs11031006 | 30,204,981 | 8.49 × 10−14 | A > G | [23] |
| Age at menopause | rs11031006 | 30,204,981 | 8.5 × 10−14 | A > G | [31] |
| Dizygotic twinning | rs11031006 | 30,204,981 | 1.25 × 10−10 | G > A | [44] |
| FSH concentrations | rs11031005 rs11031006 | 30,204,809 30,204,981 | 1.74 × 10−8 2.3 × 10−10 | T > C G > A | [37] [44] |
| LH concentrations | rs11031002 | 30,193,714 | 3.94 × 10−9 | T > A | [37] |
| Endometriosis | rs74485684 | 30,220,740 | 2.00 × 10−8 | T > C | [27] |
| Polycystic ovarian syndrome | rs11031005 | 30,204,809 | 8.66 × 10−13 | C > T | [45] |
| Excessive, frequent and irregular menstruation | rs11031006 | 30,204,981 | 1.1 × 10−38 | A > G | [20] |
| Uterine fibroids | rs11031006 | 30,204,981 | 5.7 × 10−15 | A > G | [54] |
| Bilateral oophorectomy | rs11031006 | 30,204,981 | 1.1 × 10−38 | A > G | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGrath, I.M.; Mortlock, S.; Montgomery, G.W. Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility. Int. J. Mol. Sci. 2021, 22, 2556. https://doi.org/10.3390/ijms22052556
McGrath IM, Mortlock S, Montgomery GW. Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility. International Journal of Molecular Sciences. 2021; 22(5):2556. https://doi.org/10.3390/ijms22052556
Chicago/Turabian StyleMcGrath, Isabelle M., Sally Mortlock, and Grant W. Montgomery. 2021. "Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility" International Journal of Molecular Sciences 22, no. 5: 2556. https://doi.org/10.3390/ijms22052556
APA StyleMcGrath, I. M., Mortlock, S., & Montgomery, G. W. (2021). Genetic Regulation of Physiological Reproductive Lifespan and Female Fertility. International Journal of Molecular Sciences, 22(5), 2556. https://doi.org/10.3390/ijms22052556