
The Multifaceted Role of Plasminogen in Cancer




Abstract

{kind=link}
{kind=link}
{kind=link}
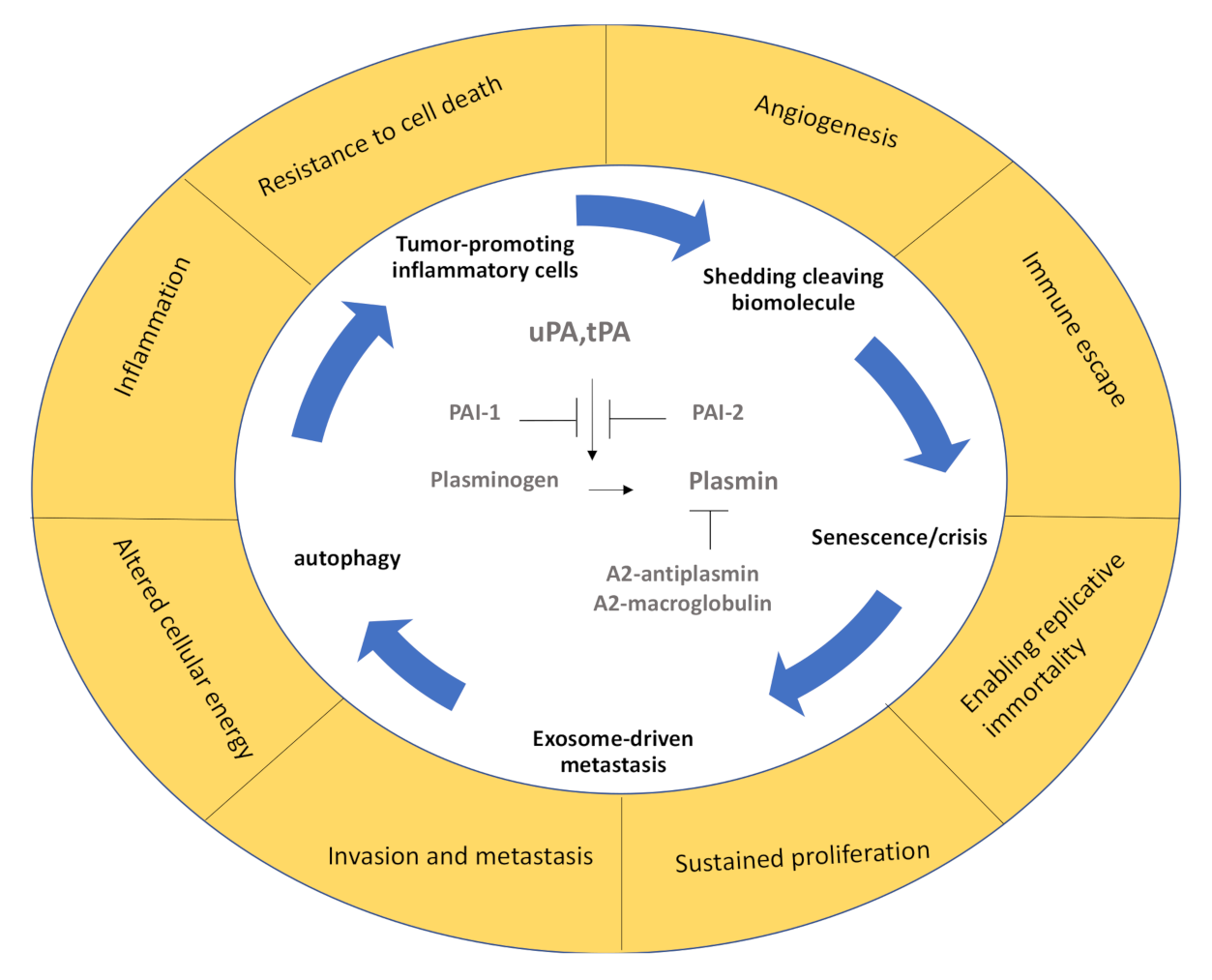
1. Introduction
2. Plasminogen Contributes to Autophagy
3. Fibrinolytic Factors and Anoikis
4. PAI-2 and Senescence
5. Fibrinolytic Factors Help to Establish the Premetastatic Niche
6. Plasminogen Receptors and Drug Resistance
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heissig, B.; Eiamboonsert, S.; Salama, Y.; Shimazu, H.; Dhahri, D.; Munakata, S.; Tashiro, Y.; Hattori, K. Cancer therapy targeting the fibrinolytic system. Adv. Drug Deliv. Rev. 2016, 99 (Pt. B), 172–179. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharm. 2018, 105, 83–94. [Google Scholar] [CrossRef]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The multifaceted role of plasminogen in inflammation. Cell Signal. 2020, 75, 109761. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Cho, Y.H.; Park, J.E.; Lim, D.S.; Lee, J.S. Tranexamic acid inhibits melanogenesis by activating the autophagy system in cultured melanoma cells. J. Dermatol. Sci. 2017, 88, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Subramanian, I.V.; Kelekar, A.; Ramakrishnan, S. Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood 2007, 109, 4793–4802. [Google Scholar] [CrossRef]
- Fang, S.; Hong, H.; Li, L.; He, D.; Xu, Z.; Zuo, S.; Han, J.; Wu, Q.; Dai, Z.; Cai, W.; et al. Plasminogen kringle 5 suppresses gastric cancer via regulating HIF-1α and GRP78. Cell Death Dis. 2017, 8, e3144. [Google Scholar] [CrossRef]
- Xiong, G.-F.; Xu, R. Function of cancer cell-derived extracellular matrix in tumor progression. J. Cancer Metastasis Treat. 2016, 2, 357–364. [Google Scholar] [CrossRef]
- Meilhac, O.; Ho-Tin-Noé, B.; Houard, X.; Philippe, M.; Michel, J.-B.; Anglés-Cano, E. Pericellular plasmin induces smooth muscle cell anoikis. Faseb J. 2003, 17, 1301–1303. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.V.; Høgdall, C.K.; Jochumsen, K.M.; Høgdall, E.V.S. Annexin A2 and cancer: A systematic review. Int. J. Oncol. 2018, 52, 5–18. [Google Scholar] [CrossRef]
- Kubala, M.H.; Punj, V.; Placencio-Hickok, V.R.; Fang, H.; Fernandez, G.E.; Sposto, R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 Promotes the Recruitment and Polarization of Macrophages in Cancer. Cell Rep. 2018, 25, 2177–2191. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends in Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Hsieh, H.-H.; Chen, Y.-C.; Jhan, J.-R.; Lin, J.-J. The serine protease inhibitor serpinB2 binds and stabilizes p21 in senescent cells. J. Cell Sci. 2017, 130, 3272. [Google Scholar] [CrossRef]
- Sossey-Alaoui, K.; Pluskota, E.; Szpak, D.; Plow, E.F. The Kindlin2-p53-SerpinB2 signaling axis is required for cellular senescence in breast cancer. Cell Death Dis. 2019, 10, 539. [Google Scholar] [CrossRef]
- Westrick, R.J.; Røjkjaer, L.P.; Yang, A.Y.; Roh, M.H.; Siebert, A.E.; Ginsburg, D. Deficiency of plasminogen activator inhibitor-2 results in accelerated tumor growth. J. Thromb. Haemost. 2020, 18, 2968–2975. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef]
- Noh, K.; Bach, D.-H.; Choi, H.-J.; Kim, M.S.; Wu, S.Y.; Pradeep, S.; Ivan, C.; Cho, M.-S.; Bayraktar, E.; Rodriguez-Aguayo, C.; et al. The hidden role of paxillin: Localization to nucleus promotes tumor angiogenesis. Oncogene 2021, 40, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Shatz-Azoulay, H.; Vinik, Y.; Isaac, R.; Kohler, U.; Lev, S.; Zick, Y. The Animal Lectin Galectin-8 Promotes Cytokine Expression and Metastatic Tumor Growth in Mice. Sci. Rep. 2020, 10, 7375. [Google Scholar] [CrossRef]
- Vinik, Y.; Shatz-Azoulay, H.; Vivanti, A.; Hever, N.; Levy, Y.; Karmona, R.; Brumfeld, V.; Baraghithy, S.; Attar-Lamdar, M.; Boura-Halfon, S.; et al. The mammalian lectin galectin-8 induces RANKL expression, osteoclastogenesis, and bone mass reduction in mice. eLife 2015, 4, e05914. [Google Scholar] [CrossRef] [PubMed]
- Roda, O.; Ortiz-Zapater, E.; Martínez-Bosch, N.; Gutiérrez-Gallego, R.; Vila-Perelló, M.; Ampurdanés, C.; Gabius, H.J.; André, S.; Andreu, D.; Real, F.X.; et al. Galectin-1 is a novel functional receptor for tissue plasminogen activator in pancreatic cancer. Gastroenterology 2009, 136, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Slatter, T.L.; Hung, N.; Campbell, H.; Rubio, C.; Mehta, R.; Renshaw, P.; Williams, G.; Wilson, M.; Engelmann, A.; Jeffs, A.; et al. Hyperproliferation, cancer, and inflammation in mice expressing a Δ133p53-like isoform. Blood 2011, 117, 5166–5177. [Google Scholar] [CrossRef]
- Cai, Q.; Dozmorov, M.; Oh, Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells 2020, 9, 1261. [Google Scholar] [CrossRef]
- Baxter, R.C.; Martin, J.L.; Beniac, V.A. High molecular weight insulin-like growth factor binding protein complex. Purification and properties of the acid-labile subunit from human serum. J. Biol. Chem. 1989, 264, 11843–11848. [Google Scholar] [CrossRef]
- Campbell, P.G.; Durham, S.K.; Suwanichkul, A.; Hayes, J.D.; Powell, D.R. Plasminogen binds the heparin-binding domain of insulin-like growth factor-binding protein-3. Am. J. Physiol. Endocrinol. Metab. 1998, 275, E321–E331. [Google Scholar] [CrossRef]
- Leal, S.M.; Liu, Q.; Huang, S.S.; Huang, J.S. The type V transforming growth factor beta receptor is the putative insulin-like growth factor-binding protein 3 receptor. J. Biol. Chem. 1997, 272, 20572–20576. [Google Scholar] [CrossRef]
- Xue, A.; Scarlett, C.J.; Jackson, C.J.; Allen, B.J.; Smith, R.C. Prognostic significance of growth factors and the urokinase-type plasminogen activator system in pancreatic ductal adenocarcinoma. Pancreas 2008, 36, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Takahashi, M.; Honke, K.; Miyoshi, E.; Osumi, D.; Sakiyama, H.; Ekuni, A.; Wang, X.; Inoue, S.; Gu, J.; et al. Loss of core fucosylation of low-density lipoprotein receptor-related protein-1 impairs its function, leading to the upregulation of serum levels of insulin-like growth factor-binding protein 3 in Fut8−/− mice. J. Biochem. 2006, 139, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Mantuano, E.; Lam, M.S.; Gonias, S.L. LRP1 assembles unique co-receptor systems to initiate cell signaling in response to tissue-type plasminogen activator and myelin-associated glycoprotein. J. Biol. Chem. 2013, 288, 34009–34018. [Google Scholar] [CrossRef]
- Zwirzitz, A.; Reiter, M.; Skrabana, R.; Ohradanova-Repic, A.; Majdic, O.; Gutekova, M.; Cehlar, O.; Petrovčíková, E.; Kutejova, E.; Stanek, G.; et al. Lactoferrin is a natural inhibitor of plasminogen activation. J. Biol. Chem. 2018, 293, 8600–8613. [Google Scholar] [CrossRef] [PubMed]
- Salama, Y.; Lin, S.Y.; Dhahri, D.; Hattori, K.; Heissig, B. The fibrinolytic factor tPA drives LRP1-mediated melanoma growth and metastasis. Faseb J. 2019, 33, 3465–3480. [Google Scholar] [CrossRef]
- Dhahri, D.; Sato-Kusubata, K.; Ohki-Koizumi, M.; Nishida, C.; Tashiro, Y.; Munakata, S.; Shimazu, H.; Salama, Y.; Eiamboonsert, S.; Nakauchi, H.; et al. Fibrinolytic crosstalk with endothelial cells expands murine mesenchymal stromal cells. Blood 2016, 128, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Dhahri, D.; Eiamboonsert, S.; Salama, Y.; Shimazu, H.; Munakata, S.; Hattori, K. Role of mesenchymal stem cell-derived fibrinolytic factor in tissue regeneration and cancer progression. Cell Mol. Life Sci. 2015, 72, 4759–4770. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Montel, V.; Gaultier, A.; Lester, R.D.; Campana, W.M.; Gonias, S.L. The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res. 2007, 67, 9817–9824. [Google Scholar] [CrossRef]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent Multi-miRNA Targeting of ApoE Drives LRP1/LRP8-Dependent Melanoma Metastasis and Angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: A role for plasminogen activation. Bmc Cancer 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal Annexin II Promotes Angiogenesis and Breast Cancer Metastasis. Mol. Cancer Res. 2017, 15, 93. [Google Scholar] [CrossRef]
- Durrieu, L.; Bharadwaj, A.; Waisman, D.M. Analysis of the thrombotic and fibrinolytic activities of tumor cell-derived extracellular vesicles. Blood Adv. 2018, 2, 1054–1065. [Google Scholar] [CrossRef]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Lund, L.R.; Akiyama, H.; Ohki, M.; Morita, Y.; Rmer, J.; Nakauchi, H.; Okumura, K.; Ogawa, H.; Werb, Z.; et al. The Plasminogen Fibrinolytic Pathway Is Required for Hematopoietic Regeneration. Cell Stem Cell 2008, 3, 120. [Google Scholar] [CrossRef]
- Fietz, T.; Hattori, K.; Thiel, E.; Heissig, B. Increased soluble urokinase plasminogen activator receptor (suPAR) serum levels after granulocyte colony-stimulating factor treatment do not predict successful progenitor cell mobilization in vivo. Blood 2006, 107, 3408–3409. [Google Scholar] [CrossRef][Green Version]
- Van Gool, B.; Dedieu, S.; Emonard, H.; Roebroek, A.J.M. The Matricellular Receptor LRP1 Forms an Interface for Signaling and Endocytosis in Modulation of the Extracellular Tumor Environment. Front. Pharmacol. 2015, 6, 271. [Google Scholar] [CrossRef]
- Gonias, S.L.; Hu, J. Urokinase receptor and resistance to targeted anticancer agents. Front. Pharmacol. 2015, 6, 154. [Google Scholar] [CrossRef]
- Zhou, J.; Kwak, K.J.; Wu, Z.; Yang, D.; Li, J.; Chang, M.; Song, Y.; Zeng, H.; Lee, L.J.; Hu, J.; et al. PLAUR Confers Resistance to Gefitinib Through EGFR/P-AKT/Survivin Signaling Pathway. Cell. Physiol. Biochem. 2018, 47, 1909–1924. [Google Scholar] [CrossRef]
- Laurenzana, A.; Margheri, F.; Biagioni, A.; Chillà, A.; Pimpinelli, N.; Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Calorini, L.; Serratì, S.; et al. EGFR/uPAR interaction as druggable target to overcome vemurafenib acquired resistance in melanoma cells. EBioMedicine 2019, 39, 194–206. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Leslie, P.L.; Franklin, D.A.; Liu, Y.; Zhang, Y. p53 Regulates the Expression of LRP1 and Apoptosis through a Stress Intensity-Dependent MicroRNA Feedback Loop. Cell Rep. 2018, 24, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, R.A.; Liu, Q.; Kanekiyo, T.; Zhang, J.; Bu, G. Low density lipoprotein receptor-related protein 1 promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J. Biol. Chem. 2009, 284, 34045–34053. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heissig, B.; Salama, Y.; Osada, T.; Okumura, K.; Hattori, K. The Multifaceted Role of Plasminogen in Cancer. Int. J. Mol. Sci. 2021, 22, 2304. https://doi.org/10.3390/ijms22052304
Heissig B, Salama Y, Osada T, Okumura K, Hattori K. The Multifaceted Role of Plasminogen in Cancer. International Journal of Molecular Sciences. 2021; 22(5):2304. https://doi.org/10.3390/ijms22052304
Chicago/Turabian StyleHeissig, Beate, Yousef Salama, Taro Osada, Ko Okumura, and Koichi Hattori. 2021. "The Multifaceted Role of Plasminogen in Cancer" International Journal of Molecular Sciences 22, no. 5: 2304. https://doi.org/10.3390/ijms22052304
APA StyleHeissig, B., Salama, Y., Osada, T., Okumura, K., & Hattori, K. (2021). The Multifaceted Role of Plasminogen in Cancer. International Journal of Molecular Sciences, 22(5), 2304. https://doi.org/10.3390/ijms22052304