
Contribution of Particle-Induced Lysosomal Membrane Hyperpolarization to Lysosomal Membrane Permeabilization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
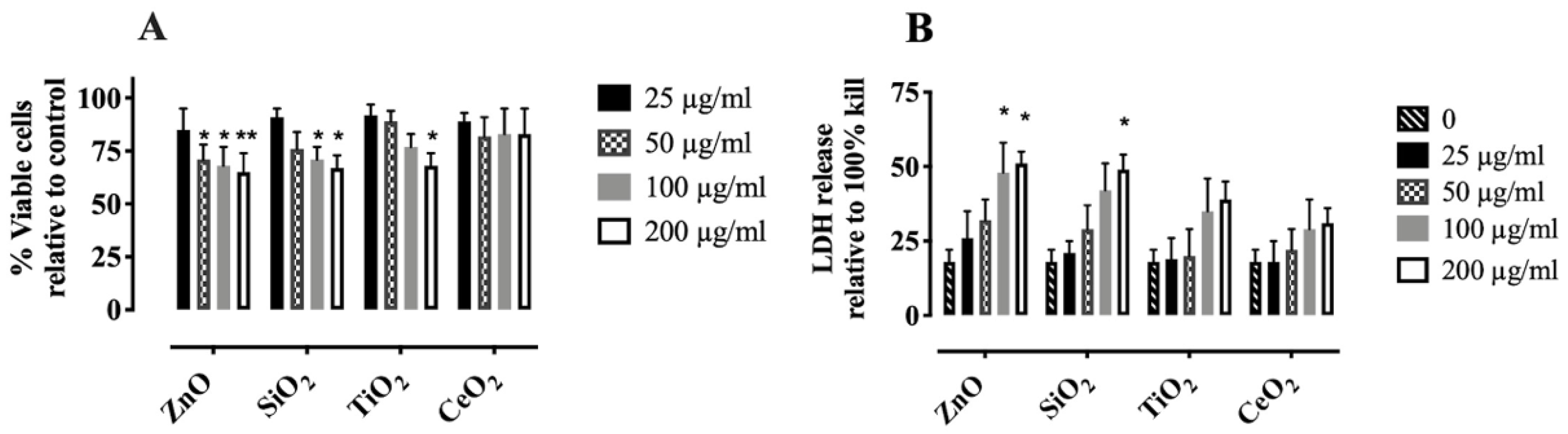
2.1. Characterization and Cytotoxicity of NP and SiO2
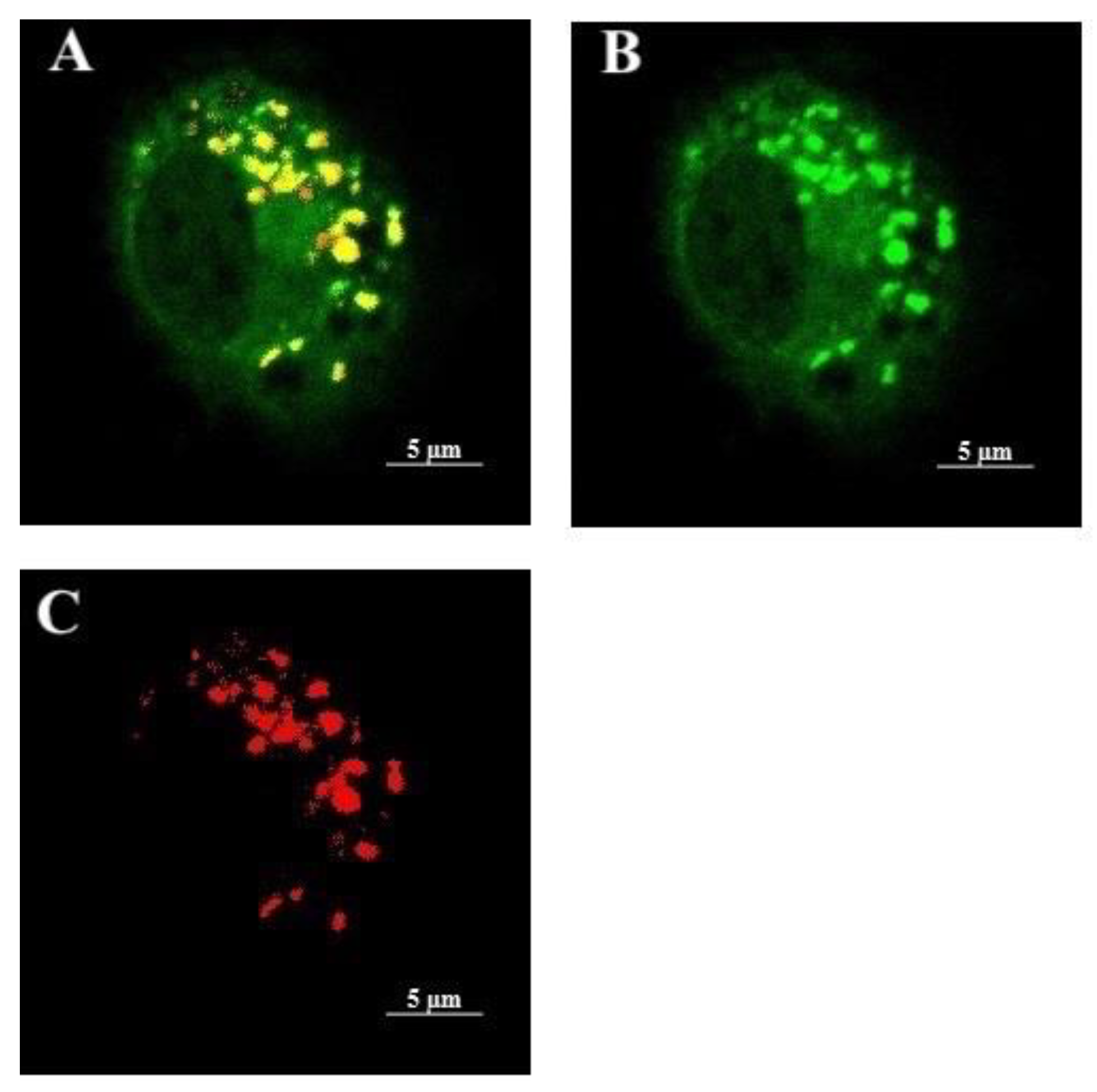
2.2. Validation of Lysosomal Membrane Hyperpolarization Using DiBAC4(3)
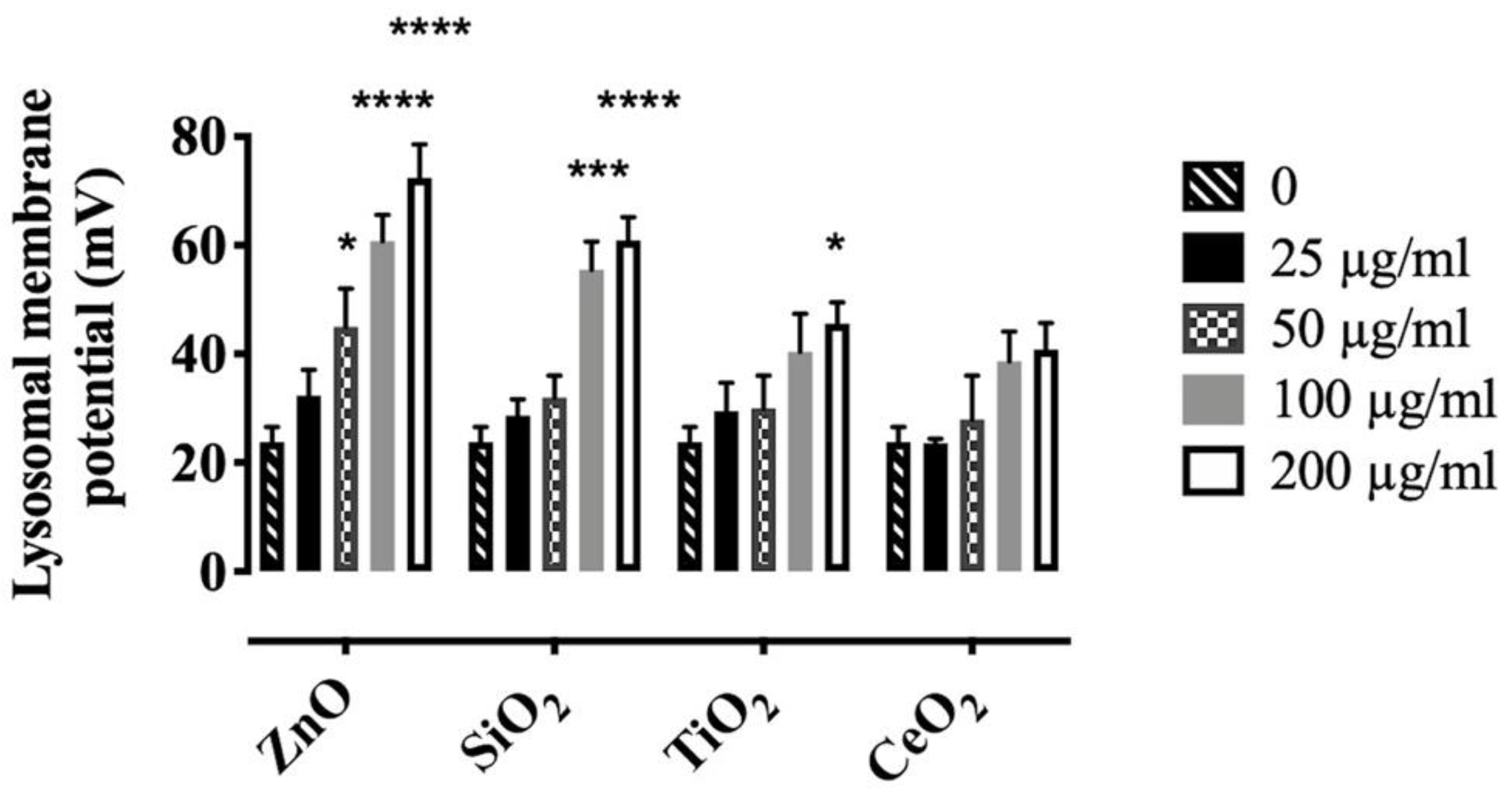
2.3. Effects of NP and SiO2 on Lysosomal Membrane Potential
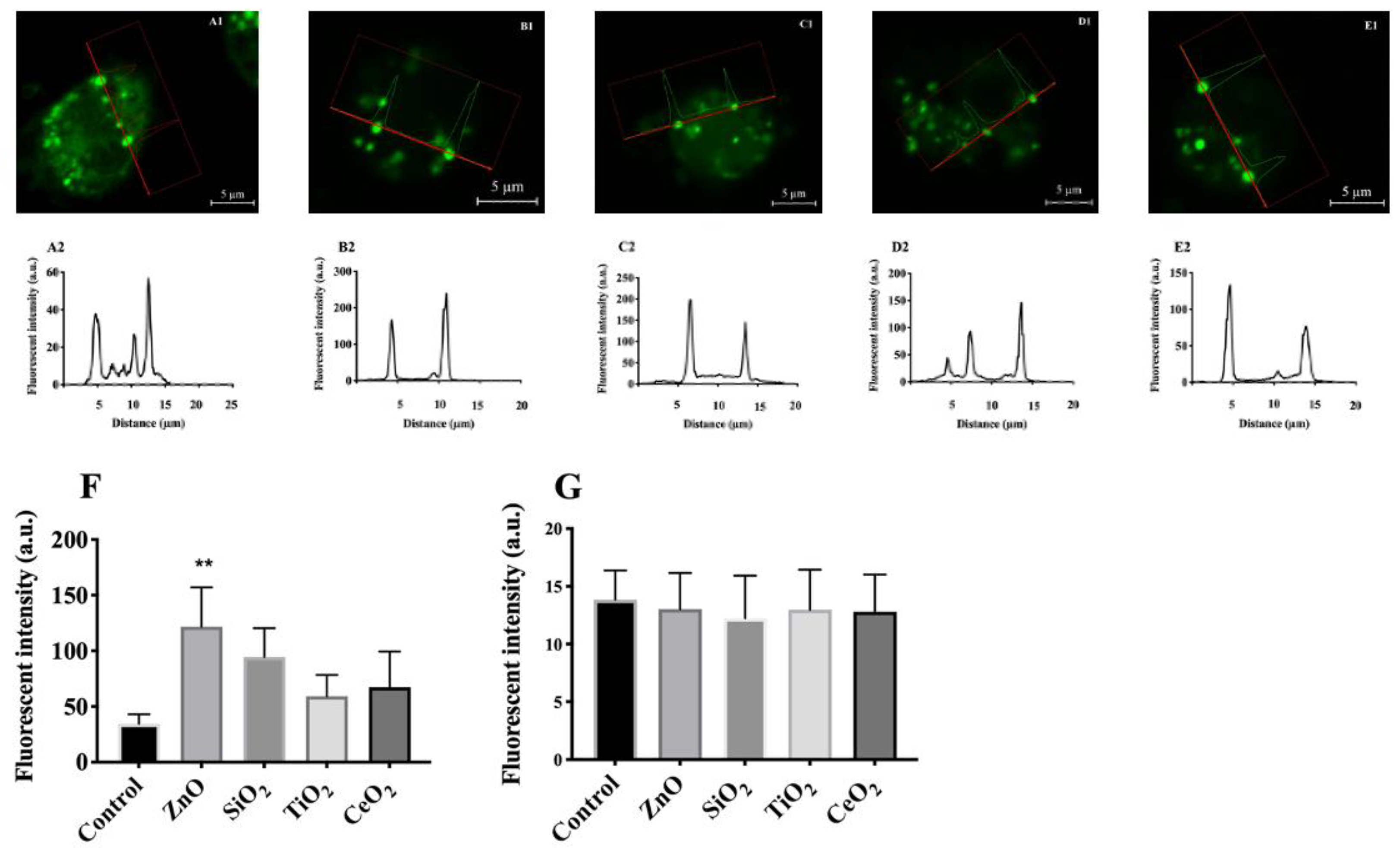
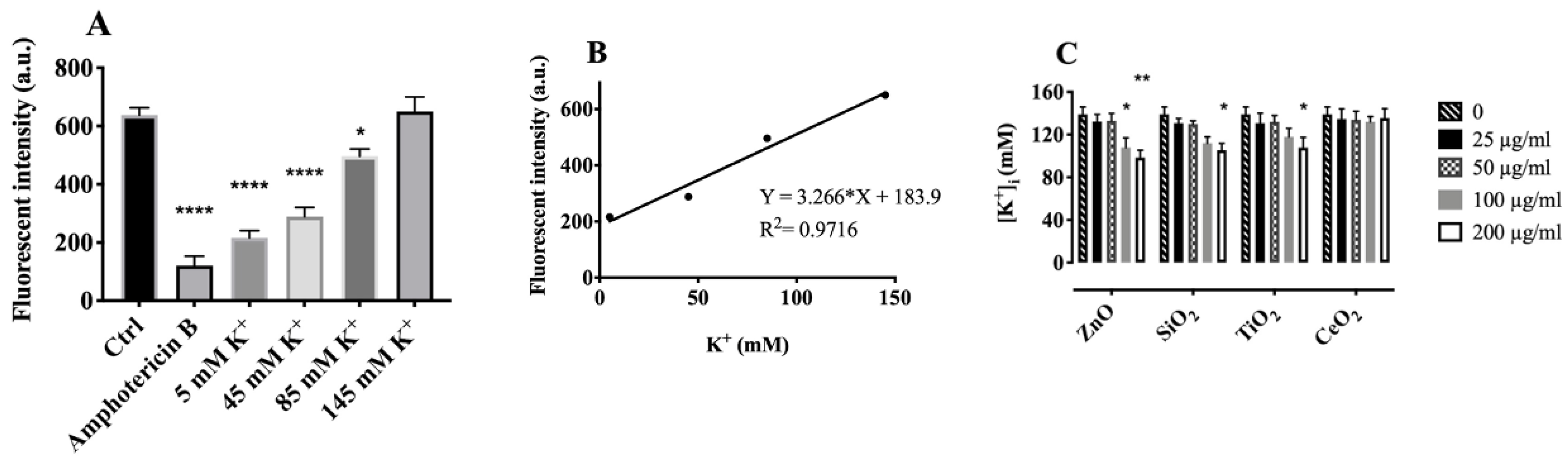
2.4. Effects of Particles on Cytosolic Potassium [K+]
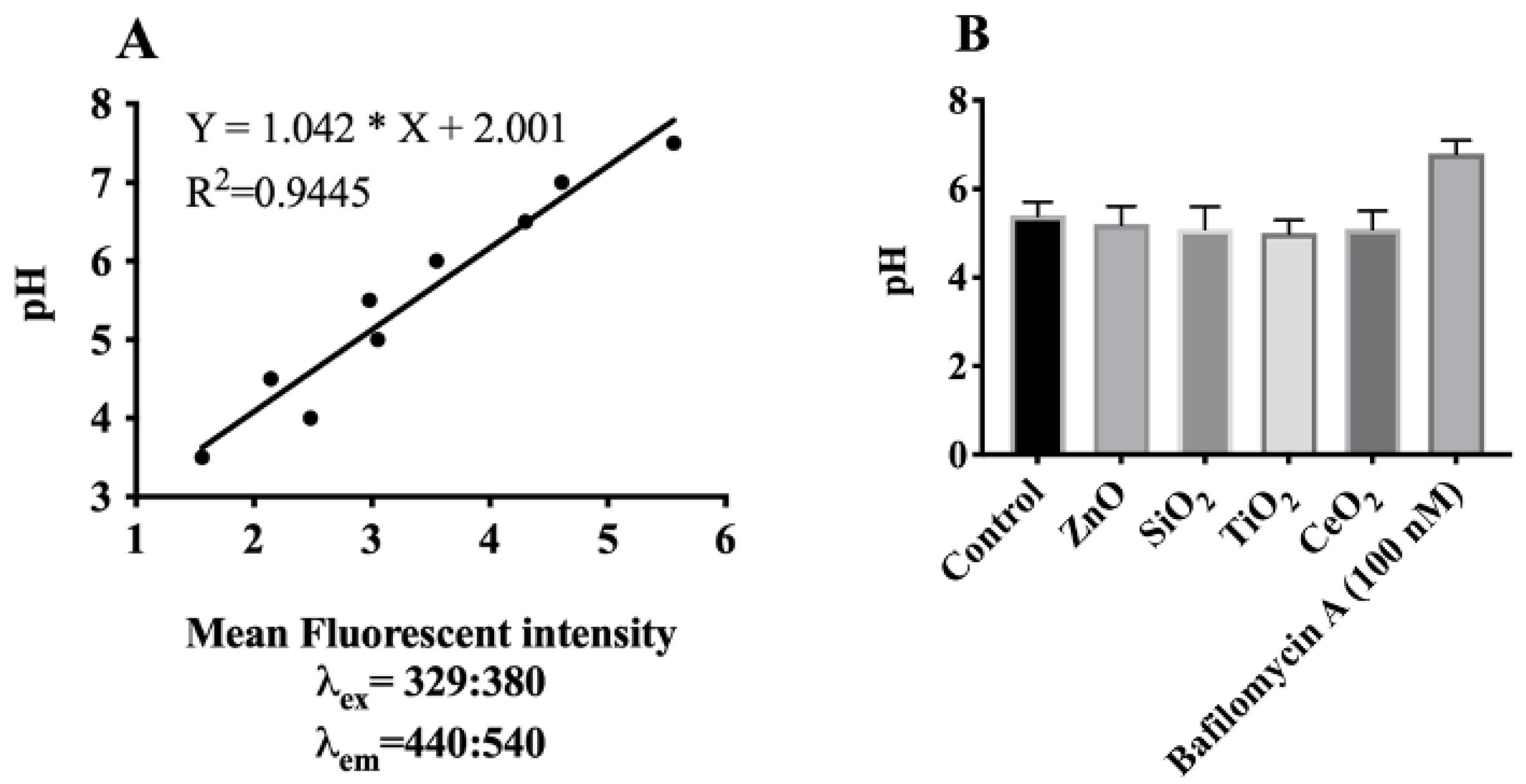
2.5. Effects of Lysosomal H+-Influx on Lysosomal Hyperpolarization
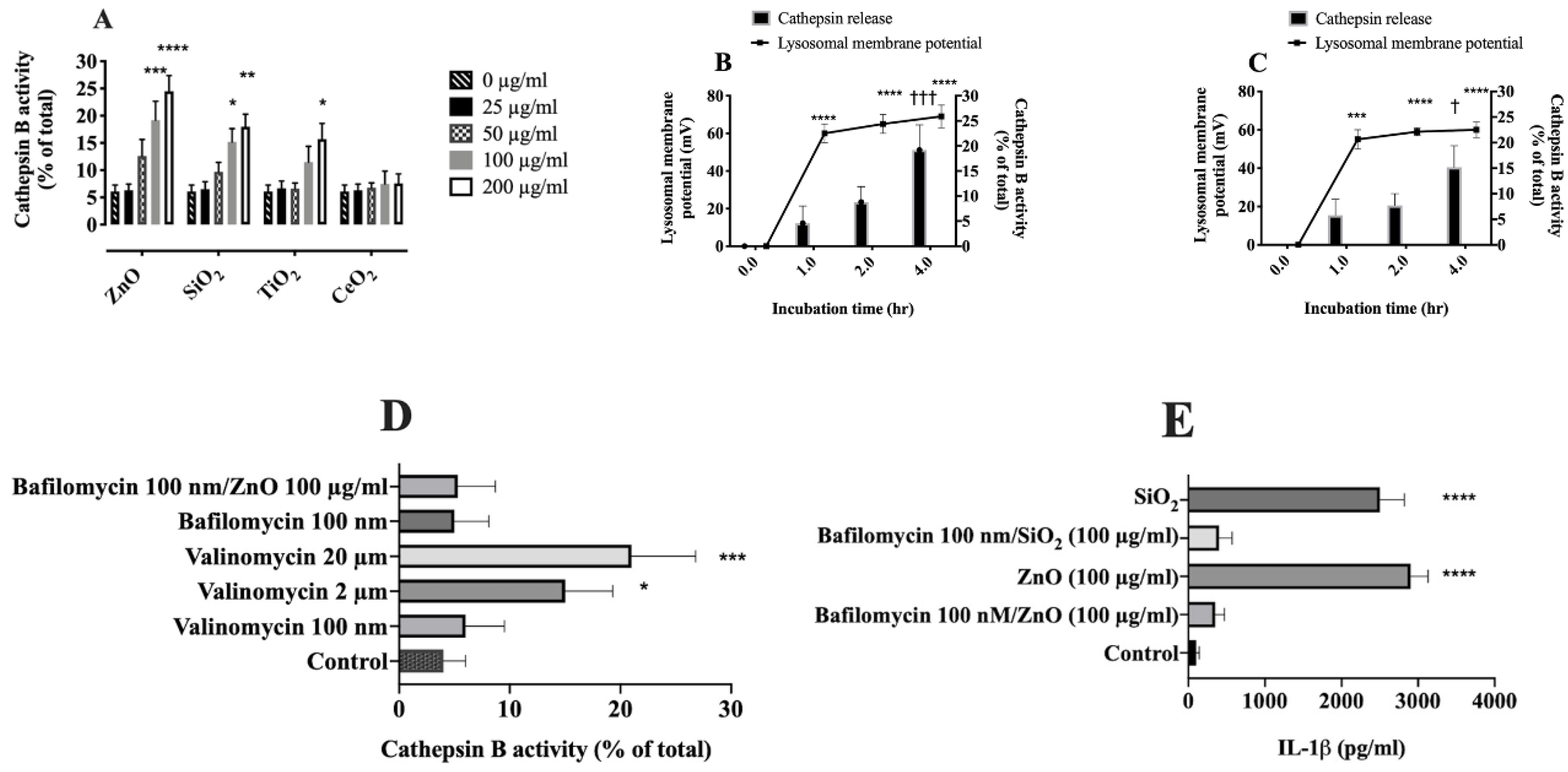
2.6. Relationship between Lysosomal Hyperpolarization, LMP, and NLRP3 Inflammasome Activity
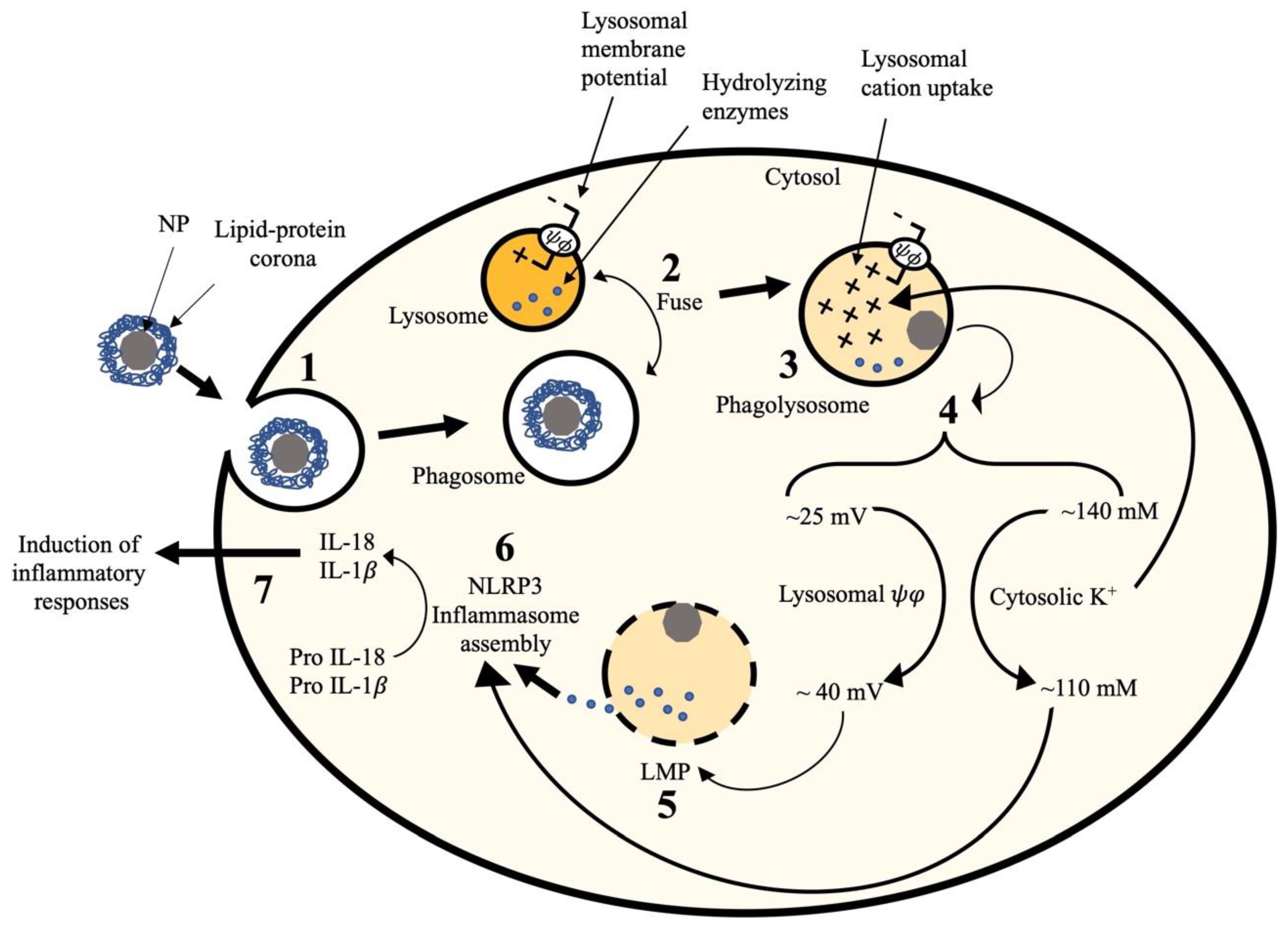
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Particles
4.3. Preparation of NP and SiO2 in Cell Culture Media
4.4. Mice
4.5. Alveolar Macrophages
4.6. Preparation of Small Unilamellar Vesicles
4.7. Lysosomal Membrane Potential Measurement
4.8. Plasma Membrane Potential Measurement
4.9. Cytosolic [K+] Measurement
4.10. Measurement of Lysosomal H+
4.11. Lysosomal Membrane Permeabilization Assay
4.12. Transmission Electron Microscopy Imaging
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitrano, D.M.; Motellier, S.; Clavaguera, S.; Nowack, B. Review of nanomaterial aging and transformations through the life cycle of nano-enhanced products. Environ. Int. 2015, 77, 132–147. [Google Scholar] [CrossRef]
- Hochella, M.F., Jr.; Mogk, D.W.; Ranville, J.; Allen, I.C.; Luther, G.W.; Marr, L.C.; McGrail, B.P.; Murayama, M.; Qafoku, N.P.; Rosso, K.M.; et al. Natural, incidental, and engineered nanomaterials and their impacts on the Earth system. Science 2019, 363, eaau8299. [Google Scholar] [CrossRef]
- Bowman, D.M.; van Calster, G.; Friedrichs, S. Nanomaterials and regulation of cosmetics. Nat. Nanotechnol. 2010, 5, 2. [Google Scholar] [CrossRef]
- Fadeel, B.; Farcal, L.; Hardy, B.; Vazquez-Campos, S.; Hristozov, D.; Marcomini, A.; Lynch, I.; Valsami-Jones, E.; Alenius, H.; Savolainen, K. Advanced tools for the safety assessment of nanomaterials. Nat. Nanotechnol. 2018, 13, 537–543. [Google Scholar] [CrossRef]
- Xia, T.; Hamilton, R.F.; Bonner, J.C.; Crandall, E.D.; Elder, A.; Fazlollahi, F.; Girtsman, T.A.; Kim, K.; Mitra, S.; Ntim, S.A.; et al. Interlaboratory evaluation of in vitro cytotoxicity and inflammatory responses to engineered nanomaterials: The NIEHS Nano GO Consortium. Environ. Health Perspect. 2013, 121, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Tang, M. The inflammatory response to silver and titanium dioxide nanoparticles in the central nervous system. Nanomedicine 2018, 13, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.P.; Guan, R.F.; Tao, M.; Lyu, F.; Cao, G.Z.; Liu, M.Q.; Gao, J.G. Assessment of the toxicity and inflammatory effects of different-sized zinc oxide nanoparticles in 2D and 3D cell cultures. Rsc. Adv. 2017, 7, 12437–12445. [Google Scholar] [CrossRef]
- Song, W.J.; Jeong, M.S.; Choi, D.M.; Kim, K.N.; Wie, M.B. Zinc Oxide Nanoparticles Induce Autophagy and Apoptosis via Oxidative Injury and Pro-Inflammatory Cytokines in Primary Astrocyte Cultures. Nanomaterials 2019, 9, 1043. [Google Scholar] [CrossRef]
- Ray, J.L.; Fletcher, P.; Burmeister, R.; Holian, A. The role of sex in particle-induced inflammation and injury. Wiley Interdiscip Rev. Nanomed. Nanobiotechnol. 2019, 12, e1589. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, R.; Hueber, A.J.; Hutton, A.; McInnes, I.B.; Graham, D. Nanoparticles and Inflammation. Thescientificworldjo 2011, 11, 1300–1312. [Google Scholar] [CrossRef]
- Bunderson-Schelvan, M.; Holian, A.; Hamilton, R.F., Jr. Engineered nanomaterial-induced lysosomal membrane permeabilization and anti-cathepsin agents. J. Toxicol. Environ. Health B Crit. Rev. 2017, 20, 230–248. [Google Scholar] [CrossRef]
- Ono, H.; Ohta, R.; Kawasaki, Y.; Niwa, A.; Takada, H.; Nakahata, T.; Ohga, S.; Saito, M.K. Lysosomal membrane permeabilization causes secretion of IL-1beta in human vascular smooth muscle cells. Inflamm. Res. 2018, 67, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglie, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef]
- Biswas, R.; Hamilton, R.F., Jr.; Holian, A. Role of lysosomes in silica-induced inflammasome activation and inflammation in absence of MARCO. J. Immunol. Res. 2014, 2014, 304180. [Google Scholar] [CrossRef]
- Murschhauser, A.; Rottgermann, P.J.F.; Woschee, D.; Ober, M.F.; Yan, Y.; Dawson, K.A.; Radler, J.O. A high-throughput microscopy method for single-cell analysis of event-time correlations in nanoparticle-induced cell death. Commun. Biol. 2019, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhang, G.J. Lysosomal destabilization via increased potassium ion permeability following photodamage. Biochim. Biophys. Acta 1997, 1323, 334–342. [Google Scholar] [CrossRef][Green Version]
- Zhang, G.; Yi, Y.P.; Zhang, G.J. Effects of arachidonic acid on the lysosomal ion permeability and osmotic stability. J. Bioenerg. Biomembr. 2006, 38, 75–82. [Google Scholar] [CrossRef]
- Deng, D.; Jiang, N.; Hao, S.J.; Sun, H.; Zhang, G.J. Loss of membrane cholesterol influences lysosomal permeability to potassium ions and protons. Biochim. Biophys. Acta 2009, 1788, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhu, M.X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China Life Sci. 2016, 59, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Steinberg, B.E.; Mason, D.; Grinstein, S. In situ measurement of the electrical potential across the lysosomal membrane using FRET. Traffic 2011, 12, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Piccinno, F.; Gottschalk, F.; Seeger, S.; Nowack, B. Industrial production quantities and uses of ten engineered nanomaterials in Europe and the world. J. Nanoparticle Res. 2012, 14, 1109. [Google Scholar] [CrossRef]
- Hamilton, R.F., Jr.; Thakur, S.A.; Holian, A. Silica binding and toxicity in alveolar macrophages. Free Radic. Biol. Med. 2008, 44, 1246–1258. [Google Scholar] [CrossRef]
- Sager, T.M.; Wolfarth, M.; Leonard, S.S.; Morris, A.M.; Porter, D.W.; Castranova, V.; Holian, A. Role of engineered metal oxide nanoparticle agglomeration in reactive oxygen species generation and cathepsin B release in NLRP3 inflammasome activation and pulmonary toxicity. Inhal. Toxicol. 2016, 28, 686–697. [Google Scholar] [CrossRef]
- Jessop, F.; Hamilton, R.F., Jr.; Rhoderick, J.F.; Fletcher, P.; Holian, A. Phagolysosome acidification is required for silica and engineered nanoparticle-induced lysosome membrane permeabilization and resultant NLRP3 inflammasome activity. Toxicol. Appl. Pharm. 2017, 318, 58–68. [Google Scholar] [CrossRef]
- Ziglari, T.; Anderson, D.; Holian, A. Determination of the relative contribution of the non-dissolved fraction of ZnO NP on membrane permeability and cytotoxicity. Inhal. Toxicol. 2020, in press. [Google Scholar] [CrossRef]
- Adams, D.S.; Levin, M. Measuring resting membrane potential using the fluorescent voltage reporters DiBAC4(3) and CC2-DMPE. Cold Spring Harb. Protoc. 2012, 2012, 459–464. [Google Scholar] [CrossRef]
- Klapperstuck, T.; Glanz, D.; Hanitsch, S.; Klapperstuck, M.; Markwardt, F.; Wohlrab, J. Calibration procedures for the quantitative determination of membrane potential in human cells using anionic dyes. Cytom. A 2013, 83, 612–626. [Google Scholar] [CrossRef]
- Warren, E.A.; Payne, C.K. Cellular binding of nanoparticles disrupts the membrane potential. Rsc. Adv. 2015, 5, 13660–13666. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. Calcium Signaling: From Basic to Bedside. Adv. Exp. Med. Biol. 2020, 1131, 1–6. [Google Scholar]
- Wang, W.; Zhang, X.; Gao, Q.; Lawas, M.; Yu, L.; Cheng, X.; Gu, M.; Sahoo, N.; Li, X.; Li, P.; et al. A voltage-dependent K(+) channel in the lysosome is required for refilling lysosomal Ca(2+) stores. J. Cell. Biol. 2017, 216, 1715–1730. [Google Scholar] [CrossRef]
- Klemm, A.R.; Pell, K.L.; Anderson, L.M.; Andrew, C.L.; Lloyd, J.B. Lysosome membrane permeability to anions. Biochim. Biophys. Acta 1998, 1373, 17–26. [Google Scholar] [CrossRef][Green Version]
- Porter, J.; Pickup, R.; Edwards, C. Membrane Hyperpolarization by Valinomycin and Its Limitations for Bacterial Viability Assessment Using Rhodamine-123 and Flow-Cytometry. FEMS Microbiol. Lett. 1995, 132, 259–262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rana, P.S.; Gibbons, B.A.; Vereninov, A.A.; Yurinskaya, V.E.; Clements, R.J.; Model, T.A.; Model, M.A. Calibration and characterization of intracellular Asante Potassium Green probes, APG-2 and APG-4. Anal. Biochem. 2019, 567, 8–13. [Google Scholar] [CrossRef]
- Rimmele, T.S.; Chatton, J.Y. A novel optical intracellular imaging approach for potassium dynamics in astrocytes. PLoS ONE 2014, 9, e109243. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef]
- Ma, L.; Ouyang, Q.; Werthmann, G.C.; Thompson, H.M.; Morrow, E.M. Live-cell Microscopy and Fluorescence-based Measurement of Luminal pH in Intracellular Organelles. Front. Cell. Dev. Biol. 2017, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jaattela, M.; Nylandsted, J. Methods for the quantification of lysosomal membrane permeabilization: A hallmark of lysosomal cell death. Methods Cell. Biol. 2015, 126, 261–285. [Google Scholar]
- Furlong, I.J.; Lopez Mediavilla, C.; Ascaso, R.; Lopez Rivas, A.; Collins, M.K. Induction of apoptosis by valinomycin: Mitochondrial permeability transition causes intracellular acidification. Cell Death Differ. 1998, 5, 214–221. [Google Scholar] [CrossRef]
- Klein, B.; Worndl, K.; Lutz-Meindl, U.; Kerschbaum, H.H. Perturbation of intracellular K(+) homeostasis with valinomycin promotes cell death by mitochondrial swelling and autophagic processes. Apoptosis 2011, 16, 1101–1117. [Google Scholar] [CrossRef]
- Reijngoud, D.J.; Tager, J.M. The permeability properties of the lysosomal membrane. Biochim. Biophys. Acta 1977, 472, 419–449. [Google Scholar] [CrossRef]
- Henning, R. pH gradient across the lysosomal membrane generated by selective cation permeability and Donnan equilibrium. Biochim. Biophys. Acta 1975, 401, 307–316. [Google Scholar] [CrossRef]
- Casey, R.P.; Hollemans, M.; Tager, J.M. The permeability of the lysosomal membrane to small ions. Biochim. Biophys. Acta 1978, 508, 15–26. [Google Scholar] [CrossRef]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef]
- Ruth, R.C.; Weglicki, W.B. Effects of ATP on lysosomes: Protection against hyperosmolar KCl. Am. J. Physiol. 1983, 245, C68–C73. [Google Scholar] [CrossRef] [PubMed]
- Sydor, M.J.; Anderson, D.S.; Steele, H.B.B.; Ross, J.B.A.; Holian, A. Effects of titanium dioxide and zinc oxide nano-materials on lipid order in model membranes. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183313. [Google Scholar] [CrossRef]
- Hamilton, R.F., Jr.; Thakur, S.A.; Mayfair, J.K.; Holian, A. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J. Biol. Chem. 2006, 281, 34218–34226. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.J.; Hwang, L.Y.; Solandt, J.J.R.; Frank, C.W. Comparison of Extruded and Sonicated Vesicles for Planar Bilayer Self-Assembly. Materials 2013, 6, 3294–3308. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Parmryd, I. Quantifying colocalization by correlation: The Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytom. A 2010, 77, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.F.; Wu, Z.; Mitra, S.; Holian, A. The Effects of Varying Degree of MWCNT Carboxylation on Bioactivity in Various In Vivo and In Vitro Exposure Models. Int. J. Mol. Sci. 2018, 19, 354. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziglari, T.; Wang, Z.; Holian, A. Contribution of Particle-Induced Lysosomal Membrane Hyperpolarization to Lysosomal Membrane Permeabilization. Int. J. Mol. Sci. 2021, 22, 2277. https://doi.org/10.3390/ijms22052277
Ziglari T, Wang Z, Holian A. Contribution of Particle-Induced Lysosomal Membrane Hyperpolarization to Lysosomal Membrane Permeabilization. International Journal of Molecular Sciences. 2021; 22(5):2277. https://doi.org/10.3390/ijms22052277
Chicago/Turabian StyleZiglari, Tahereh, Zifan Wang, and Andrij Holian. 2021. "Contribution of Particle-Induced Lysosomal Membrane Hyperpolarization to Lysosomal Membrane Permeabilization" International Journal of Molecular Sciences 22, no. 5: 2277. https://doi.org/10.3390/ijms22052277
APA StyleZiglari, T., Wang, Z., & Holian, A. (2021). Contribution of Particle-Induced Lysosomal Membrane Hyperpolarization to Lysosomal Membrane Permeabilization. International Journal of Molecular Sciences, 22(5), 2277. https://doi.org/10.3390/ijms22052277