
Potential Neuroprotective Mechanisms of Methamphetamine Treatment in Traumatic Brain Injury Defined by Large-Scale IonStar-Based Quantitative Proteomics

 , and
, and 
Abstract
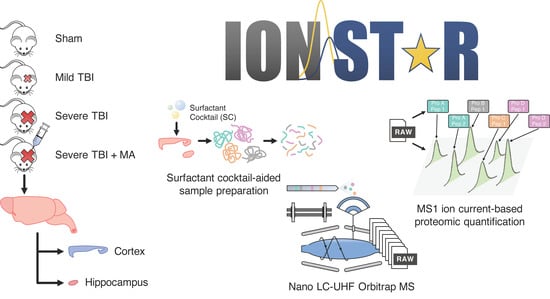
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. In-Depth Characterization of Rat Brain Proteome Using IonStar
2.2. Comparison of Global Protein Changes Post TBI and MA Treatment
2.3. Classification of APs into MA-Induced and MA-Unique Subgroups
3. Discussion
3.1. Global Patterns of Alterations in Neuroproteomics
3.2. Alteration in Canonical Pathways
3.3. Unique MA-Mediated Alterations to Upstream Regulators
3.4. Alterations in Specific Protein Clusters
4. Materials and Methods
4.1. Animal Experiments
4.2. Protein Extraction and Digestion
4.3. Nano LC-Orbitrap MS Analysis
4.4. Protein Identification and Quantification
4.5. Data Analysis and Bioinformatics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | Altered proteins |
| BCA | Bicinchoninic acid assay |
| BP | Biological processes |
| DTT | Dithiothreitol |
| EN | Experimental Null |
| FA | Formic acid |
| FADR | False Altered-protein Discovery Rate |
| FC | Fold change |
| FPI | Fluid percussion injury |
| GO | Gene Ontology |
| IAM | Iodoacetamide |
| IPA | Ingenuity Pathway Analysis |
| LC | Liquid chromatography |
| MA | Methamphetamine |
| MS | Mass spectrometry |
| NSS | Neurological Severity Score |
| PCA | Principal Component Analysis |
| PPI | Protein–protein interaction |
| ROS | Reactive oxygen species |
| SEPOD | Surfactant cocktail-aided Extraction/Precipitation/On-pellet Digestion |
| SH | Sham control |
| TBI | Traumatic brain injury |
| TM | Mild TBI |
| TS | Severe TBI |
| TSm | Severe TBI with MA treatment |
| UHF | Ultra-high-field |
References
- Centers for Disease Control and Prevention. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; National Center for Injury Prevention and Control, Division of Unintentional Injury Prevention: Atlanta, GA, USA, 2015.
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315. [Google Scholar] [CrossRef]
- McConeghy, K.W.; Hatton, J.; Hughes, L.; Cook, A.M. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 2012, 26, 613–636. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.H.; Guingab-Cagmat, J.D.; Zhang, Z.; Moghieb, A.; Glushakova, O.Y.; Mondello, S.; Boutté, A.M.; Anagli, J.; Rubenstein, R.; Bahmad, H.; et al. Neuroproteomics and Systems Biology Approach to Identify Temporal Biomarker Changes Post Experimental Traumatic Brain Injury in Rats. Front. Neurol. 2016, 7, 198. [Google Scholar] [CrossRef]
- Kobeissy, F.H.; Ottens, A.K.; Zhang, Z.; Liu, M.C.; Denslow, N.D.; Dave, J.R.; Tortella, F.C.; Hayes, R.L.; Wang, K.K. Novel differential neuroproteomics analysis of traumatic brain injury in rats. Mol. Cell Proteom. 2006, 5, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Mallah, K.; Quanico, J.; Raffo-Romero, A.; Cardon, T.; Aboulouard, S.; Devos, D.; Kobeissy, F.; Zibara, K.; Salzet, M.; Fournier, I. Mapping Spatiotemporal Microproteomics Landscape in Experimental Model of Traumatic Brain Injury Unveils a link to Parkinson’s Disease. Mol. Cell Proteom. 2019, 18, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Fang, S.; Gao, J.; Wang, J.; Cao, Z.; Guo, Z.; Huang, Q.; Qu, Y.; Zhou, H.; Yu, J. Quantitative Proteomic Study Reveals Up-Regulation of cAMP Signaling Pathway-Related Proteins in Mild Traumatic Brain Injury. J. Proteome Res. 2018, 17, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhao, Y.; Haidacher, S.J.; Wang, E.; Parsley, M.O.; Gao, J.; Sadygov, R.G.; Starkey, J.M.; Luxon, B.A.; Spratt, H.; et al. Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J. Neurotrauma 2013, 30, 775–788. [Google Scholar] [CrossRef]
- Ding, G.L.; Chopp, M.; Poulsen, D.J.; Li, L.; Qu, C.; Li, Q.; Nejad-Davarani, S.P.; Budaj, J.S.; Wu, H.; Mahmood, A.; et al. MRI of neuronal recovery after low-dose methamphetamine treatment of traumatic brain injury in rats. PLoS ONE 2013, 8, e61241. [Google Scholar] [CrossRef]
- Rau, T.F.; Kothiwal, A.S.; Rova, A.R.; Brooks, D.M.; Poulsen, D.J. Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. J. Trauma Acute Care Surg. 2012, 73 (Suppl. 1), S165–S172. [Google Scholar] [CrossRef]
- Rau, T.F.; Kothiwal, A.; Zhang, L.; Ulatowski, S.; Jacobson, S.; Brooks, D.M.; Cardozo-Pelaez, F.; Chopp, M.; Poulsen, D.J. Low dose methamphetamine mediates neuroprotection through a PI3K-AKT pathway. Neuropharmacology 2011, 61, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Rau, T.F.; Kothiwal, A.S.; Rova, A.R.; Brooks, D.M.; Rhoderick, J.F.; Poulsen, A.J.; Hutchinson, J.; Poulsen, D.J. Administration of low dose methamphetamine 12 h after a severe traumatic brain injury prevents neurological dysfunction and cognitive impairment in rats. Exp. Neurol. 2014, 253, 31–40. [Google Scholar] [CrossRef]
- Shen, S.; Wang, X.; Orsburn, B.C.; Qu, J. How could IonStar challenge the current status quo of quantitative proteomics in large sample cohorts? Expert Rev. Proteom. 2018, 15, 541–543. [Google Scholar] [CrossRef]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Poulsen, D.J.; Orsburn, B.C.; Wang, J.; et al. IonStar enables high-precision, low-missing-data proteomics quantification in large biological cohorts. Proc. Natl. Acad. Sci. USA 2018, 115, E4767–E4776. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Hu, Q.; Li, J.; Wang, J.; Qu, J. Experimental Null Method to Guide the Development of Technical Procedures and to Control False-Positive Discovery in Quantitative Proteomics. J. Proteome Res. 2015, 14, 4147–4157. [Google Scholar] [CrossRef]
- Pascovici, D.; Handler, D.C.; Wu, J.X.; Haynes, P.A. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 2016, 16, 2448–2453. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.D.; Wohlgehagen, E.; Rau, T.; Poulsen, D. Temporal and Spatial Changes in the Pattern of Iba1 and CD68 Staining in the Rat Brain Following Severe Traumatic Brain Injury. Mod. Res. Inflamm. 2015, 4, 9–23. [Google Scholar] [CrossRef]
- Toffolo, K.; Osei, J.; Kelly, W.; Poulsen, A.; Donahue, K.; Wang, J.; Hunter, M.; Bard, J.; Wang, J.; Poulsen, D. Circulating microRNAs as biomarkers in traumatic brain injury. Neuropharmacology 2019, 145 Pt B, 199–208. [Google Scholar] [CrossRef]
- Fang, X.X.; Jiang, X.L.; Han, X.H.; Peng, Y.P.; Qiu, Y.H. Neuroprotection of interleukin-6 against NMDA-induced neurotoxicity is mediated by JAK/STAT3, MAPK/ERK, and PI3K/AKT signaling pathways. Cell Mol. Neurobiol. 2013, 33, 241–251. [Google Scholar] [CrossRef]
- Liu, Z.; Qiu, Y.H.; Li, B.; Ma, S.H.; Peng, Y.P. Neuroprotection of interleukin-6 against NMDA-induced apoptosis and its signal-transduction mechanisms. Neurotox. Res. 2011, 19, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Peng, Y.P.; Lu, J.H.; Cao, B.B.; Qiu, Y.H. Neuroprotection of interleukin-6 against NMDA attack and its signal transduction by JAK and MAPK. Neurosci. Lett. 2009, 450, 122–126. [Google Scholar] [CrossRef]
- Winter, C.D.; Pringle, A.K.; Clough, G.F.; Church, M.K. Raised parenchymal interleukin-6 levels correlate with improved outcome after traumatic brain injury. Brain 2004, 127 Pt 2, 315–320. [Google Scholar] [CrossRef]
- Divolis, G.; Stavropoulos, A.; Manioudaki, M.; Apostolidou, A.; Doulou, A.; Gavriil, A.; Dafnis, I.; Chroni, A.; Mummery, C.; Xilouri, M.; et al. Activation of both transforming growth factor-beta and bone morphogenetic protein signalling pathways upon traumatic brain injury restrains pro-inflammatory and boosts tissue reparatory responses of reactive astrocytes and microglia. Brain Commun. 2019, 1, fcz028. [Google Scholar] [CrossRef]
- Hewett, S.J.; Jackman, N.A.; Claycomb, R.J. Interleukin-1beta in Central Nervous System Injury and Repair. Eur. J. Neurodegener Dis. 2012, 1, 195–211. [Google Scholar] [PubMed]
- Song, C.; Zhang, Y.; Dong, Y. Acute and subacute IL-1beta administrations differentially modulate neuroimmune and neurotrophic systems: Possible implications for neuroprotection and neurodegeneration. J. Neuroinflamm. 2013, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Li, P.; Ning, Y.L.; Peng, Y.; Xiong, R.P.; Yang, N.; Chen, X.; Zhou, Y.G. Adenosine A2A receptor inhibition restores the normal transport of endothelial glutamate transporters in the brain. Biochem. Biophys. Res. Commun. 2018, 498, 795–802. [Google Scholar] [CrossRef]
- Ning, C.; Wen, J.; Zhang, Y.; Dai, Y.; Wang, W.; Zhang, W.; Qi, L.; Grenz, A.; Eltzschig, H.K.; Blackburn, M.R.; et al. Excess adenosine A2B receptor signaling contributes to priapism through HIF-1alpha mediated reduction of PDE5 gene expression. FASEB J. 2014, 28, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.A.; Zhao, Y.; Ning, Y.L.; Yang, N.; Peng, Y.; Li, P.; Chen, X.Y.; Liu, D.; Wang, H.; Chen, X.; et al. Adenosine A2A receptor inactivation alleviates early-onset cognitive dysfunction after traumatic brain injury involving an inhibition of tau hyperphosphorylation. Transl. Psychiatry 2017, 7, e1123. [Google Scholar] [CrossRef] [PubMed]
- Laroche, M.; Kutcher, M.E.; Huang, M.C.; Cohen, M.J.; Manley, G.T. Coagulopathy after traumatic brain injury. Neurosurgery 2012, 70, 1334–1345. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Gu, Z.; Wang, D.; Wang, T.; Wang, Z.; Tao, L. Increased expression of calcium/calmodulin-dependent protein kinase type II subunit delta after rat traumatic brain injury. J. Mol. Neurosci. 2012, 46, 631–643. [Google Scholar] [CrossRef]
- Samuels, J.M.; Moore, E.E.; Silliman, C.C.; Banerjee, A.; Cohen, M.J.; Ghasabyan, A.; Chandler, J.; Coleman, J.R.; Sauaia, A. Severe traumatic brain injury is associated with a unique coagulopathy phenotype. J. Trauma Acute Care Surg. 2019, 86, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Johnson, K.J.; Murozono, M.; Sakai, K.; Magnuson, M.A.; Wieloch, T.; Cronberg, T.; Isshiki, A.; Erickson, H.P.; Fassler, R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 2001, 7, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.C.; Garcia, A.J.; LaPlaca, M.C. Plasma fibronectin is neuroprotective following traumatic brain injury. Exp. Neurol. 2007, 207, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.R.; Jenkins, T.M.; Addington, C.P.; Stabenfeldt, S.E.; Lifshitz, J. Extracellular matrix proteins are time-dependent and regional-specific markers in experimental diffuse brain injury. Brain Behav. 2020, 10, e01767. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; An, B.; Wang, X.; Hilchey, S.P.; Li, J.; Cao, J.; Tian, Y.; Hu, C.; Jin, L.; Ng, A.; et al. Surfactant Cocktail-Aided Extraction/Precipitation/On-Pellet Digestion Strategy Enables Efficient and Reproducible Sample Preparation for Large-Scale Quantitative Proteomics. Anal. Chem. 2018, 90, 10350–10359. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Shen, S.; Li, J.; Hu, Q.; Nie, L.; Tu, C.; Wang, X.; Orsburn, B.; Wang, J.; Qu, J. An IonStar Experimental Strategy for MS1 Ion Current-Based Quantification Using Ultrahigh-Field Orbitrap: Reproducible, In-Depth, and Accurate Protein Measurement in Large Cohorts. J. Proteome Res. 2017, 16, 2445–2456. [Google Scholar] [CrossRef]
- Sadygov, R.G.; Maroto, F.M.; Hühmer, A.F. ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC-MS chromatographic surfaces. Anal. Chem. 2006, 78, 8207–8217. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, S.; Zhang, M.; Ma, M.; Rasam, S.; Poulsen, D.; Qu, J. Potential Neuroprotective Mechanisms of Methamphetamine Treatment in Traumatic Brain Injury Defined by Large-Scale IonStar-Based Quantitative Proteomics. Int. J. Mol. Sci. 2021, 22, 2246. https://doi.org/10.3390/ijms22052246
Shen S, Zhang M, Ma M, Rasam S, Poulsen D, Qu J. Potential Neuroprotective Mechanisms of Methamphetamine Treatment in Traumatic Brain Injury Defined by Large-Scale IonStar-Based Quantitative Proteomics. International Journal of Molecular Sciences. 2021; 22(5):2246. https://doi.org/10.3390/ijms22052246
Chicago/Turabian StyleShen, Shichen, Ming Zhang, Min Ma, Sailee Rasam, David Poulsen, and Jun Qu. 2021. "Potential Neuroprotective Mechanisms of Methamphetamine Treatment in Traumatic Brain Injury Defined by Large-Scale IonStar-Based Quantitative Proteomics" International Journal of Molecular Sciences 22, no. 5: 2246. https://doi.org/10.3390/ijms22052246
APA StyleShen, S., Zhang, M., Ma, M., Rasam, S., Poulsen, D., & Qu, J. (2021). Potential Neuroprotective Mechanisms of Methamphetamine Treatment in Traumatic Brain Injury Defined by Large-Scale IonStar-Based Quantitative Proteomics. International Journal of Molecular Sciences, 22(5), 2246. https://doi.org/10.3390/ijms22052246