
Structural Communication between the E. coli Chaperones DnaK and Hsp90


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Measured Quantities
2.1.1. Structural Perturbation Method (SPM)
2.1.2. Preparation of Structures
3. Results
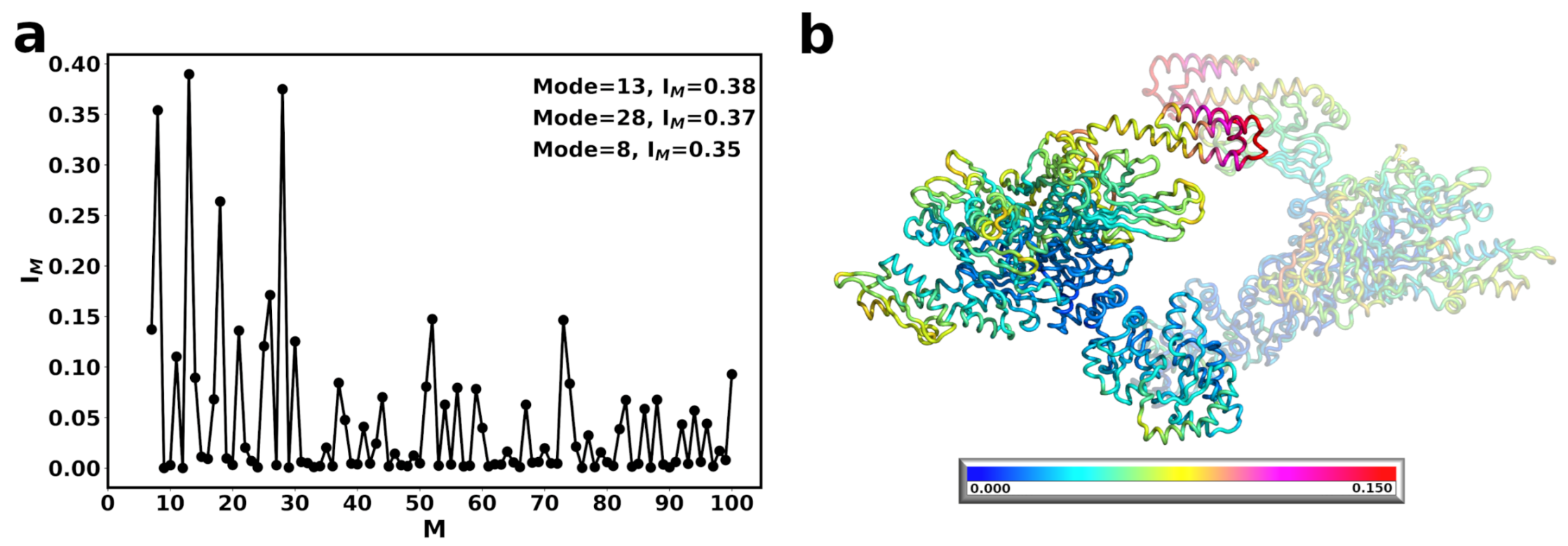
3.1. The Conformational Transition of Hsp90 from the Open → Close Conformation Can Be Described by Multiple Normal Modes
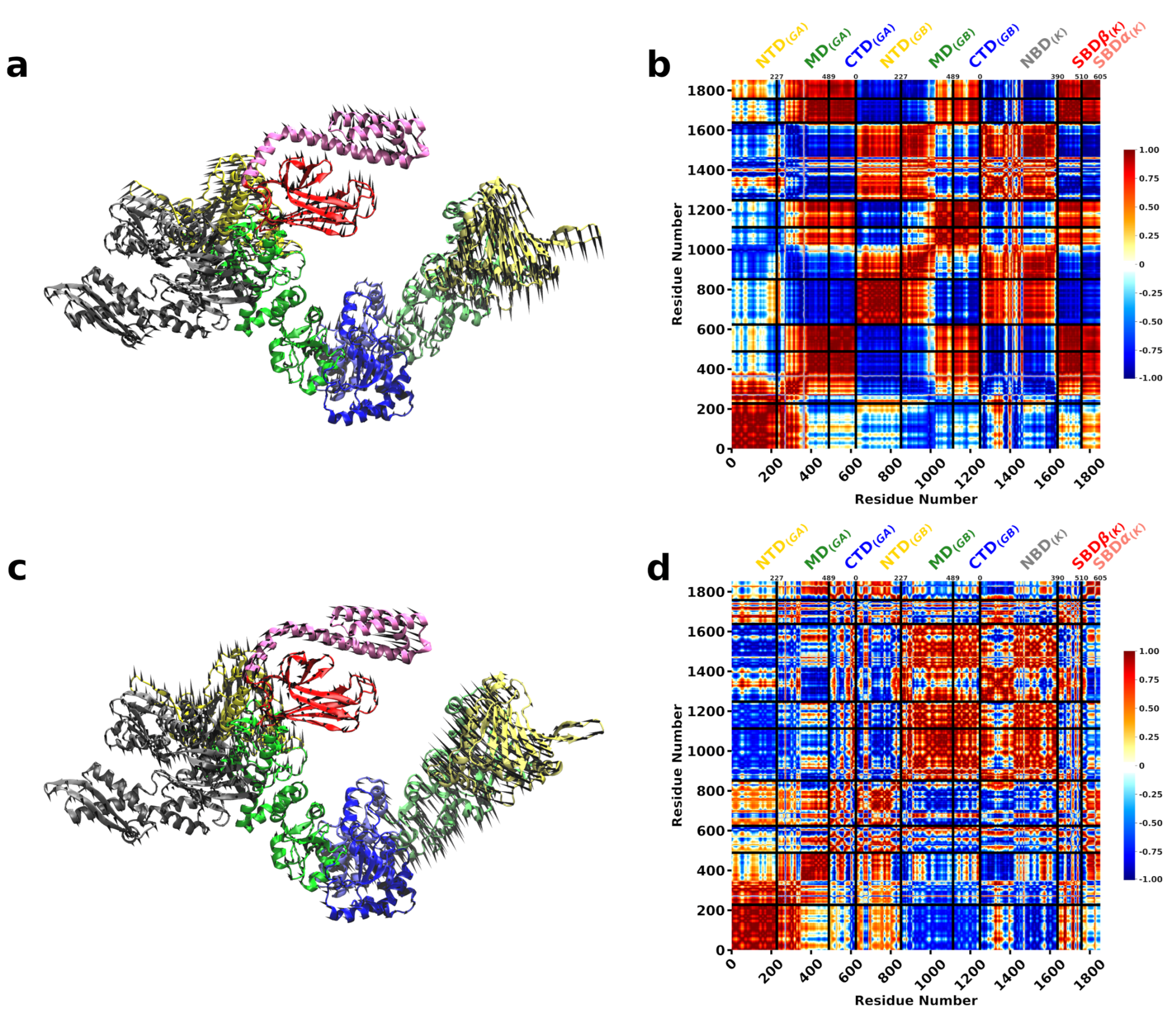
3.2. A Single DnaK Molecule Modulates the Conformational Flexibility of the Bound Hsp90 Protomer
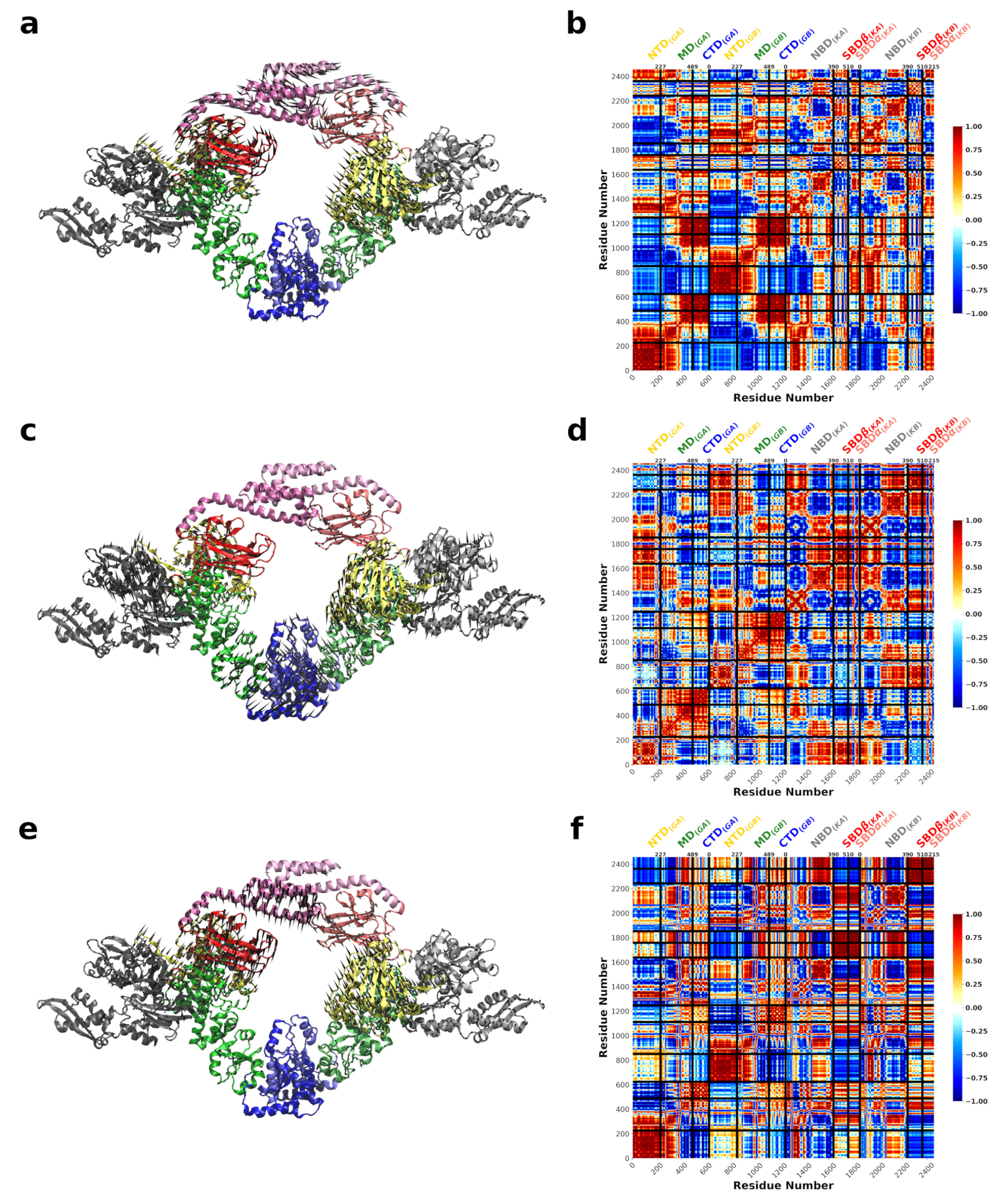
3.3. DnaK-Hsp90 Stoichiometry of 1:1 Returns the Conformational Flexibility and Symmetry within Hsp90
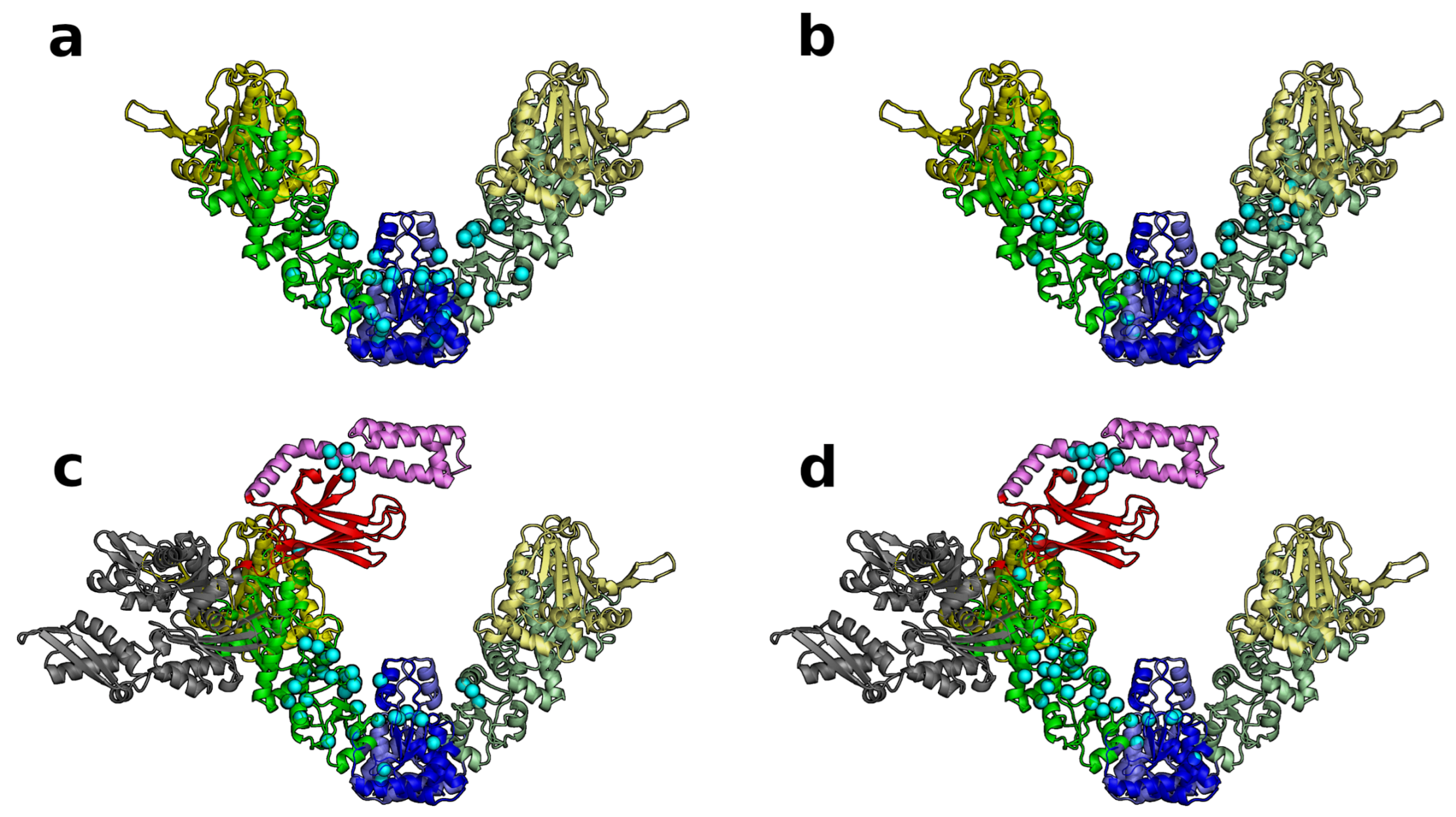
3.4. SPM Analysis Reveals a Change in the Allosteric Wiring Network When DnaK Is Bound
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SBD | substrate binding domain |
| NBD | nucleotide binding domain |
| HtpG | Hsp90 |
| DnaK | Hsp70 |
| client | substrate |
| NTD | N-terminal domain |
| MD | middle domain |
| CTD | C-terminal domain |
| ENM | Elastic Network Model |
| NMA | Normal Mode Analysis |
| SPM | Structural Perturbation Method |
References
- Wiech, H.; Buchner, J.; Zimmermann, R.; Jakob, U. Hsp90 chaperones protein folding in vitro. Nature 1992, 358, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.M.; Shastry, S.; Kravats, A.N.; Shih, Y.H.; Miot, M.; Hoskins, J.R.; Stan, G.; Wickner, S. Interplay between E. coli DnaK, ClpB and GrpE during Protein Disaggregation. J. Mol. Biol. 2015, 427, 312–327. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Bagatell, R.; Falsey, R. The Stress Response: Implications for the Clinical Development of Hsp90 Inhibitors. Curr. Cancer Drug Targets 2003, 3, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A Quantitative Chaperone Interaction Network Reveals the Architecture of Cellular Protein Homeostasis Pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef]
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the Symmetry. Mol. Cell 2015, 58, 8–20. [Google Scholar] [CrossRef]
- Röhl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237.e4. [Google Scholar] [CrossRef] [PubMed]
- Morán Luengo, T.; Kityk, R.; Mayer, M.P.; Rüdiger, S.G.D. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell 2018, 70, 545–552.e9. [Google Scholar] [CrossRef] [PubMed]
- Wegele, H.; Wandinger, S.K.; Schmid, A.B.; Reinstein, J.; Buchner, J. Substrate Transfer from the Chaperone Hsp70 to Hsp90. J. Mol. Biol. 2006, 356, 802–811. [Google Scholar] [CrossRef]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef]
- Genest, O.; Hoskins, J.R.; Camberg, J.L.; Doyle, S.M.; Wickner, S. Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling. Proc. Natl. Acad. Sci. USA 2011, 108, 8206–8211. [Google Scholar] [CrossRef]
- Richter, K.; Buchner, J. Hsp90: Chaperoning signal transduction. J. Cell. Physiol. 2001, 188, 281–290. [Google Scholar] [CrossRef]
- Marcu, M.G.; Chadli, A.; Bouhouche, I.; Catelli, M.; Neckers, L.M. The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-binding Domain in the Carboxyl Terminus of the Chaperone. J. Biol. Chem. 2000, 275, 37181–37186. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.A.; Wang, R.Y.R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Picard, D.; Khursheed, B.; Garabedian, M.J.; Fortin, M.G.; Lindquist, S.; Yamamoto, K.R. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature 1990, 348, 166–168. [Google Scholar] [CrossRef]
- Murphy, P.J.; Kanelakis, K.C.; Galigniana, M.D.; Morishima, Y.; Pratt, W.B. Stoichiometry, abundance, and functional significance of the hsp90/hsp70-based multiprotein chaperone machinery in reticulocyte lysate. J. Biol. Chem. 2001, 276, 30092–30098. [Google Scholar] [CrossRef]
- Pratt, W.B.; Galigniana, M.D.; Harrell, J.M.; DeFranco, D.B. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell. Signal. 2004, 16, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, O.R.; Freiburger, L.; Rutz, D.A.; Krause, M.; Zierer, B.K.; Alvira, S.; Cuéllar, J.; Valpuesta, J.M.; Madl, T.; Sattler, M.; et al. Modulation of the Hsp90 Chaperone Cycle by a Stringent Client Protein. Mol. Cell 2014, 53, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, S.; Freund, S.M.V.; Veprintsev, D.B.; Fersht, A.R. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc. Natl. Acad. Sci. USA 2002, 99, 11085–11090. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.M.; Westlake, C.A.; Gasiewicz, T.A. Epigallocatechin Gallate Inhibits Aryl Hydrocarbon Receptor Gene Transcription through an Indirect Mechanism Involving Binding to a 90 kDa Heat Shock Protein. Biochemistry 2005, 44, 5041–5052. [Google Scholar] [CrossRef]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In Vivo Function of Hsp90 Is Dependent on ATP Binding and ATP Hydrolysis. J. Cell Biol. 1998, 143, 901–910. [Google Scholar] [CrossRef]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed]
- Southworth, D.R.; Agard, D.A. Species-Dependent Ensembles of Conserved Conformational States Define the Hsp90 Chaperone ATPase Cycle. Mol. Cell 2008, 32, 631–640. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Böttcher, U.M.K.; Southworth, D.R.; Agard, D.A. Grp94, the endoplasmic reticulum Hsp90, has a similar solution conformation to cytosolic Hsp90 in the absence of nucleotide. Protein Sci. 2009, 18, 1815–1827. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Southworth, D.R.; Street, T.O.; Agard, D.A. pH-Dependent Conformational Changes in Bacterial Hsp90 Reveal a Grp94-Like Conformation at pH 6 That Is Highly Active in Suppression of Citrate Synthase Aggregation. J. Mol. Biol. 2009, 390, 278–291. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural Analysis of E. coli hsp90 Reveals Dramatic Nucleotide-Dependent Conformational Rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Chen, B.; Zhong, D.; Monteiro, A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genom. 2006, 7, 156. [Google Scholar] [CrossRef]
- Frey, S.; Leskovar, A.; Reinstein, J.; Buchner, J. The ATPase Cycle of the Endoplasmic Chaperone Grp94. J. Biol. Chem. 2007, 282, 35612–35620. [Google Scholar] [CrossRef]
- Schulze, J.O.; Saladino, G.; Busschots, K.; Neimanis, S.; Süß, E.; Odadzic, D.; Zeuzem, S.; Hindie, V.; Herbrand, A.K.; Lisa, M.N.; et al. Bidirectional Allosteric Communication between the ATP-Binding Site and the Regulatory PIF Pocket in PDK1 Protein Kinase. Cell Chem. Biol. 2016, 23, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Ratzke, C.; Hellenkamp, B.; Hugel, T. Four-colour FRET reveals directionality in the Hsp90 multicomponent machinery. Nat. Commun. 2014, 5, 4192. [Google Scholar] [CrossRef] [PubMed]
- Giannoulis, A.; Feintuch, A.; Barak, Y.; Mazal, H.; Albeck, S.; Unger, T.; Yang, F.; Su, X.C.; Goldfarb, D. Two closed ATP- and ADP-dependent conformations in yeast Hsp90 chaperone detected by Mn(II) EPR spectroscopic techniques. Proc. Natl. Acad. Sci. USA 2020, 117, 395–404. [Google Scholar] [CrossRef]
- Bardwell, J.C.; Craig, E.A. Ancient heat shock gene is dispensable. J. Bacteriol. 1988, 170, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.G.; Baneyx, F. Roles of the Escherichia coli Small Heat Shock Proteins IbpA and IbpB in Thermal Stress Management: Comparison with ClpA, ClpB, and HtpG In Vivo. J. Bacteriol. 1998, 180, 5165–5172. [Google Scholar] [CrossRef]
- Thomas, J.G.; Baneyx, F. ClpB and HtpG facilitate de novo protein folding in stressed Escherichia coli cells. Mol. Microbiol. 2000, 36, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop Modulates hsp70/hsp90 Interactions in Protein Folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef]
- Felts, S.J.; Toft, D.O. p23, a simple protein with complex activities. Cell Stress Chaperones 2003, 8, 108–113. [Google Scholar] [CrossRef]
- Nadeau, K.; Das, A.; Walsh, C.T. Hsp90 chaperonins possess ATPase activity and bind heat shock transcription factors and peptidyl prolyl isomerases. J. Biol. Chem. 1993, 268, 1479–1487. [Google Scholar] [CrossRef]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, S.; Piper, P.W.; Pearl, L.H. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef]
- Prodromou, C. The ‘active life’ of Hsp90 complexes. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 614–623. [Google Scholar] [CrossRef]
- Onuoha, S.C.; Coulstock, E.T.; Grossmann, J.G.; Jackson, S.E. Structural Studies on the Co-chaperone Hop and Its Complexes with Hsp90. J. Mol. Biol. 2008, 379, 732–744. [Google Scholar] [CrossRef]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 Chaperone Ligands Control Domain Association via an Allosteric Mechanism Mediated by the Interdomain Linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Yang, J.; Wong, J.L.; Vorvis, C.; Hendrickson, W.A.; et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef]
- Szabo, A.; Langer, T.; Schroder, H.; Flanagan, J.; Bukau, B.; Hartl, F.U. The ATP Hydrolysis-Dependent Reaction Cycle of the Escherichia coli Hsp70 System-DnaK, DnaJ, and GrpE. Proc. Natl. Acad. Sci. USA 1994, 91, 10345–10349. [Google Scholar] [CrossRef]
- Lewis, M.; Pelham, H. Involvement of ATP in the nuclear and nucleolar functions of the 70 kd heat shock protein. EMBO J. 1985, 4, 3137–3143. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J. Proteins as molecular chaperones. Nature 1987, 328, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Karzai, A.W.; McMacken, R. A Bipartite Signaling Mechanism Involved in DnaJ-mediated Activation of the Escherichia coli DnaK Protein. J. Biol. Chem. 1996, 271, 11236–11246. [Google Scholar] [CrossRef]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef]
- Rauch, J.N.; Gestwicki, J.E. Binding of Human Nucleotide Exchange Factors to Heat Shock Protein 70 (Hsp70) Generates Functionally Distinct Complexes in Vitro. J. Biol. Chem. 2014, 289, 1402–1414. [Google Scholar] [CrossRef]
- Raviol, H.; Sadlish, H.; Rodriguez, F.; Mayer, M.P.; Bukau, B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006, 25, 2510–2518. [Google Scholar] [CrossRef]
- Dragovic, Z.; Broadley, S.A.; Shomura, Y.; Bracher, A.; Hartl, F.U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006, 25, 2519–2528. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 Chaperone Machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef]
- Nathan, D.F.; Vos, M.H.; Lindquist, S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Natl. Acad. Sci. USA 1997, 94, 12949–12956. [Google Scholar] [CrossRef] [PubMed]
- Picard, D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. CMLS 2002, 59, 1640–1648. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the Chaperone Network: An Integrative Map of Physical and Genetic Interactions Mediated by the Hsp90 Chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Nakamoto, H.; Fujita, K.; Ohtaki, A.; Watanabe, S.; Narumi, S.; Maruyama, T.; Suenaga, E.; Misono, T.S.; Kumar, P.K.R.; Goloubinoff, P.; et al. Physical interaction between bacterial heat shock protein (Hsp) 90 and Hsp70 chaperones mediates their cooperative action to refold denatured proteins. J. Biol. Chem. 2014, 289, 6110–6119. [Google Scholar] [CrossRef] [PubMed]
- Genest, O.; Hoskins, J.R.; Kravats, A.N.; Doyle, S.M.; Wickner, S. Hsp70 and Hsp90 of E. coli Directly Interact for Collaboration in Protein Remodeling. J. Mol. Biol. 2015, 427, 3877–3889. [Google Scholar] [CrossRef]
- Kravats, A.N.; Doyle, S.M.; Hoskins, J.R.; Genest, O.; Doody, E.; Wickner, S. Interaction of E. coli Hsp90 with DnaK Involves the DnaJ Binding Region of DnaK. J. Mol. Biol. 2017, 429, 858–872. [Google Scholar] [CrossRef] [PubMed]
- Kravats, A.N.; Hoskins, J.R.; Reidy, M.; Johnson, J.L.; Doyle, S.M.; Genest, O.; Masison, D.C.; Wickner, S. Functional and physical interaction between yeast Hsp90 and Hsp70. Proc. Natl. Acad. Sci. USA 2018, 115, E2210–E2219. [Google Scholar] [CrossRef]
- Sung, N.; Lee, J.; Kim, J.H.; Chang, C.; Joachimiak, A.; Lee, S.; Tsai, F.T.F. Mitochondrial Hsp90 is a ligand-activated molecular chaperone coupling ATP binding to dimer closure through a coiled-coil intermediate. Proc. Natl. Acad. Sci. USA 2016, 113, 2952–2957. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kotler, J.L.M.; Liu, S.; Street, T.O. The endoplasmic reticulum (ER) chaperones BiP and Grp94 selectively associate when BiP is in the ADP conformation. J. Biol. Chem. 2019, 294, 6387–6396. [Google Scholar] [CrossRef]
- Doyle, S.M.; Hoskins, J.R.; Kravats, A.N.; Heffner, A.L.; Garikapati, S.; Wickner, S. Intermolecular Interactions between Hsp90 and Hsp70. J. Mol. Biol. 2019, 431, 2729–2746. [Google Scholar] [CrossRef]
- Genest, O.; Reidy, M.; Street, T.O.; Hoskins, J.R.; Camberg, J.L.; Agard, D.A.; Masison, D.C.; Wickner, S. Uncovering a Region of Heat Shock Protein 90 Important for Client Binding in E. coli and Chaperone Function in Yeast. Mol. Cell 2013, 49, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef]
- Pearl, L.H. The HSP90 molecular chaperone-an enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR Domain–Peptide Complexes: Critical Elements in the Assembly of the Hsp70–Hsp90 Multichaperone Machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Chen, S.; Smith, D.F. Hop as an Adaptor in the Heat Shock Protein 70 (Hsp70) and Hsp90 Chaperone Machinery. J. Biol. Chem. 1998, 273, 35194–35200. [Google Scholar] [CrossRef]
- Richter, K.; Muschler, P.; Hainzl, O.; Reinstein, J.; Buchner, J. Sti1 Is a Non-Competitive Inhibitor of the Hsp90 ATPase Binding Prevents the N-Terminal Dimerization Reaction during the ATPase Cycle. J. Biol. Chem. 2003, 278, 10328–10333. [Google Scholar] [CrossRef]
- Southworth, D.R.; Agard, D.A. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Mol. Cell 2011, 42, 771–781. [Google Scholar] [CrossRef]
- Song, G.; Jernigan, R.L. An enhanced elastic network model to represent the motions of domain-swapped proteins. Proteins Struct. Funct. Bioinform. 2006, 63, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Chennubhotla, C.; Bahar, I. Markov propagation of allosteric effects in biomolecular systems: Application to GroEL–GroES. Mol. Syst. Biol. 2006, 2, 36. [Google Scholar] [CrossRef]
- Bastolla, U. Computing protein dynamics from protein structure with elastic network models. WIREs Comput. Mol. Sci. 2014, 4, 488–503. [Google Scholar] [CrossRef]
- Kurkcuoglu, O.; Jernigan, R.L.; Doruker, P. Collective Dynamics of Large Proteins from Mixed Coarse-Grained Elastic Network Model. QSAR Comb. Sci. 2005, 24, 443–448. [Google Scholar] [CrossRef]
- Sinitskiy, A.V.; Voth, G.A. Coarse-graining of proteins based on elastic network models. Chem. Phys. 2013, 422, 165–174. [Google Scholar] [CrossRef]
- Eyal, E.; Bahar, I. Toward a Molecular Understanding of the Anisotropic Response of Proteins to External Forces: Insights from Elastic Network Models. Biophys. J. 2008, 94, 3424–3435. [Google Scholar] [CrossRef]
- Jayasinghe, M.; Shrestha, P.; Wu, X.; Tehver, R.; Stan, G. Weak Intra-Ring Allosteric Communications of the Archaeal Chaperonin Thermosome Revealed by Normal Mode Analysis. Biophys. J. 2012, 103, 1285–1295. [Google Scholar] [CrossRef]
- Tehver, R.; Chen, J.; Thirumalai, D. Allostery Wiring Diagrams in the Transitions that Drive the GroEL Reaction Cycle. J. Mol. Biol. 2009, 387, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B.R.; Doniach, S.; Thirumalai, D. Network of Dynamically Important Residues in the Open/Closed Transition in Polymerases Is Strongly Conserved. Structure 2005, 13, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Brooks, B.R. Probing the Local Dynamics of Nucleotide-Binding Pocket Coupled to the Global Dynamics: Myosin versus Kinesin. Biophys. J. 2005, 89, 167–178. [Google Scholar] [CrossRef]
- Zheng, W.; Doniach, S. A comparative study of motor-protein motions by using a simple elastic-network model. Proc. Natl. Acad. Sci. USA 2003, 100, 13253–13258. [Google Scholar] [CrossRef]
- Zheng, W.; Brooks, B.R.; Thirumalai, D. Allosteric Transitions in the Chaperonin GroEL are Captured by a Dominant Normal Mode that is Most Robust to Sequence Variations. Biophys. J. 2007, 93, 2289–2299. [Google Scholar] [CrossRef]
- Loutchko, D.; Flechsig, H. Allosteric communication in molecular machines via information exchange: What can be learned from dynamical modeling. Biophys. Rev. 2020, 12, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Sanejouand, Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. Des. Sel. 2001, 14, 1–6. [Google Scholar] [CrossRef]
- Romo, T.D.; Grossfield, A. Validating and improving elastic network models with molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2011, 79, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Villinger, S.; Gohlke, H. Large-scale comparison of protein essential dynamics from molecular dynamics simulations and coarse-grained normal mode analyses. Proteins Struct. Funct. Bioinform. 2010, 78, 3341–3352. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Bahar, I.; Flatow, D.; Covell, D.G.; Jernigan, R.L. Molecular Mechanisms of Chaperonin GroEL-GroES Function. Biochemistry 2002, 41, 491–501. [Google Scholar] [CrossRef]
- Tirion, M.M. Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys. Rev. Lett. 1996, 77, 4. [Google Scholar] [CrossRef]
- Ma, J. Usefulness and Limitations of Normal Mode Analysis in Modeling Dynamics of Biomolecular Complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Atilgan, A.; Durell, S.; Jernigan, R.; Demirel, M.; Keskin, O.; Bahar, I. Anisotropy of Fluctuation Dynamics of Proteins with an Elastic Network Model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Tehver, R.; Thirumalai, D. Rigor to post-rigor transition in myosin V: Link between the dynamics and the supporting architecture. Structure 2010, 4, 471–481. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.; Potter, S.; Finn, R.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocols 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins Struct. Funct. Bioinform. 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G.M. Probing Molecular Mechanisms of the Hsp90 Chaperone: Biophysical Modeling Identifies Key Regulators of Functional Dynamics. PLoS ONE 2012, 7, e37605. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Dancing through Life: Molecular Dynamics Simulations and Network-Centric Modeling of Allosteric Mechanisms in Hsp70 and Hsp110 Chaperone Proteins. PLoS ONE 2015, 10, e0143752. [Google Scholar] [CrossRef] [PubMed]
- Nicolaï, A.; Senet, P.; Delarue, P.; Ripoll, D.R. Human Inducible Hsp70: Structures, Dynamics, and Interdomain Communication from All-Atom Molecular Dynamics Simulations. J. Chem. Theory Comput. 2010, 6, 2501–2519. [Google Scholar] [CrossRef]
- Gołaś, E.; Maisuradze, G.G.; Senet, P.; Ołdziej, S.; Czaplewski, C.; Scheraga, H.A.; Liwo, A. Simulation of the Opening and Closing of Hsp70 Chaperones by Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 1750–1764. [Google Scholar] [CrossRef]
- Mishra, P.; Bolon, D.N.A. Designed Hsp90 Heterodimers Reveal an Asymmetric ATPase-Driven Mechanism In Vivo. Mol. Cell 2014, 53, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Lavery, L.A.; Partridge, J.R.; Ramelot, T.A.; Elnatan, D.; Kennedy, M.A.; Agard, D.A. Structural Asymmetry in the Closed State of Mitochondrial Hsp90 (TRAP1) Supports a Two-Step ATP Hydrolysis Mechanism. Mol. Cell 2014, 53, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Street, T.O.; Lavery, L.A.; Verba, K.A.; Lee, C.T.; Mayer, M.P.; Agard, D.A. Cross-Monomer Substrate Contacts Reposition the Hsp90 N-Terminal Domain and Prime the Chaperone Activity. J. Mol. Biol. 2012, 415, 3–15. [Google Scholar] [CrossRef][Green Version]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Sanchez-Martin, C.; Serapian, S.A.; Colombo, G.; Rasola, A. Dynamically Shaping Chaperones. Allosteric Modulators of HSP90 Family as Regulatory Tools of Cell Metabolism in Neoplastic Progression. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Penkler, D.; Sensoy, Ö.; Atilgan, C.; Tastan Bishop, Ö. Perturbation–Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. [Google Scholar] [CrossRef] [PubMed]
- Smock, R.G.; Rivoire, O.; Russ, W.P.; Swain, J.F.; Leibler, S.; Ranganathan, R.; Gierasch, L.M. An interdomain sector mediating allostery in Hsp70 molecular chaperones. Mol. Syst. Biol. 2010, 6. [Google Scholar] [CrossRef]
- Nicolaï, A.; Delarue, P.; Senet, P. Decipher the Mechanisms of Protein Conformational Changes Induced by Nucleotide Binding through Free-Energy Landscape Analysis: ATP Binding to Hsp70. PLoS Comput. Biol. 2013, 9, 1–20. [Google Scholar] [CrossRef]
- Liu, Y.; Bahar, I. Toward understanding allosteric signaling mechanisms in the atpase domain of molecular chaperones (Conference Paper). In Pacific Symposium on Biocomputing 2010, PSB2010; World Scientific: Kamuela, HI, USA, 2009; pp. 269–280. [Google Scholar] [CrossRef]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8. Available online: https://pymol.org/ (accessed on 20 December 2020).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The GIMP Development Team. GIMP. 2019. Available online: https://www.gimp.org (accessed on 20 December 2020).
- Inkscape Project. Inkscape. 2020. Available online: https://inkscape.org (accessed on 20 December 2020).
- Blacklock, K.; Verkhivker, G.M. Differential Modulation of Functional Dynamics and Allosteric Interactions in the Hsp90-Cochaperone Complexes with p23 and Aha1: A Computational Study. PLoS ONE 2013, 8, e71936. [Google Scholar] [CrossRef]
- Morra, G.; Verkhivker, G.; Colombo, G. Modeling Signal Propagation Mechanisms and Ligand- Based Conformational Dynamics of the Hsp90 Molecular Chaperone Full-Length Dimer. PLoS Comput. Biol. 2009, 5, 1–16. [Google Scholar] [CrossRef]
- Richter, K.; Buchner, J. hsp90: Twist and Fold. Cell 2006, 127, 251–253. [Google Scholar] [CrossRef]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-Nucleotide Complexes Reveal Mechanistic Differences between the hsp90 Chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, M.; Elnatan, D.; Larson, A.G.; Agard, D.A. Cryo-EM analysis of human mitochondrial Hsp90 in multiple tetrameric states. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vaughan, C.K.; Mollapour, M.; Smith, J.R.; Truman, A.; Hu, B.; Good, V.M.; Panaretou, B.; Neckers, L.; Clarke, P.A.; Workman, P.; et al. Hsp90-Dependent Activation of Protein Kinases Is Regulated by Chaperone-Targeted Dephosphorylation of Cdc37. Mol. Cell 2008, 31, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Noddings, C.M.; Wang, R.Y.R.; Agard, D.A. GR chaperone cycle mechanism revealed by cryo-EM: Reactivation of GR by the GR:Hsp90:p23 client-maturation complex. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kominek, J.; Marszalek, J.; Neuvéglise, C.; Craig, E.A.; Williams, B.L. The Complex Evolutionary Dynamics of Hsp70s: A Genomic and Functional Perspective. Genome Biol. Evol. 2013, 5, 2460–2477. [Google Scholar] [CrossRef]
- Chen, C.F.; Chen, Y.; Dai, K.; Chen, P.L.; Riley, D.J.; Lee, W.H. A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol. Cell. Biol. 1996, 16, 4691–4699. [Google Scholar] [CrossRef]
- Chang, H.C.; Nathan, D.F.; Lindquist, S. In vivo analysis of the Hsp90 cochaperone Sti1 (p60). Mol. Cell. Biol. 1997, 17, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spuy, J.; Kana, B.D.; Dirr, H.W.; Blatch, G.L. Heat shock cognate protein 70 chaperone-binding site in the co-chaperone murine stress-inducible protein 1 maps to within three consecutive tetratricopeptide repeat motifs. Biochem. J. 2000, 345, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, A.M.; Cuccioloni, M.; Bellesi, J.; Lupidi, G.; Fioretti, E.; Angeletti, M. Interaction of Hsp90 with 20S proteasome: Thermodynamic and kinetic characterization. Proteins Struct. Funct. Bioinform. 2002, 48, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Jacob, J.; Michowski, W.; Nowotny, M.; Kuznicki, J.; Chazin, W.J. Human Sgt1 Binds HSP90 through the CHORD-Sgt1 Domain and Not the Tetratricopeptide Repeat Domain. J. Biol. Chem. 2004, 279, 16511–16517. [Google Scholar] [CrossRef]
- Hildenbrand, Z.L.; Molugu, S.K.; Paul, A.; Avila, G.A.; Herrera, N.; Xiao, C.; Cox, M.B.; Bernal, R.A. High-yield expression and purification of the Hsp90-associated p23, FKBP52, HOP and SGTα proteins. J. Chromatogr. B 2010, 878, 2760–2764. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, P.C.; Bernthaler, A.; Dupuis, P.; Mayer, B.; Picard, D. An Interaction Network Predicted from Public Data as a Discovery Tool: Application to the Hsp90 Molecular Chaperone Machine. PLoS ONE 2011, 6, e26044. [Google Scholar] [CrossRef]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The Plasticity of the Hsp90 Co-chaperone System. Mol. Cell 2017, 67, 947–961.e5. [Google Scholar] [CrossRef]
- Yi, F.; Doudevski, I.; Regan, L. HOP is a monomer: Investigation of the oligomeric state of the co-chaperone HOP. Protein Sci. A Publ. Protein Soc. 2010, 19, 19–25. [Google Scholar] [CrossRef][Green Version]
- Alvira, S.; Cuéllar, J.; Röhl, A.; Yamamoto, S.; Itoh, H.; Alfonso, C.; Rivas, G.; Buchner, J.; Valpuesta, J.M. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat. Commun. 2014, 5, 5484. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Graf, C.; Mayer, F.J.; Richter, S.M.; Mayer, M.P. Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 2012, 31, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.B.; Lagleder, S.; Gräwert, M.A.; Röhl, A.; Hagn, F.; Wandinger, S.K.; Cox, M.B.; Demmer, O.; Richter, K.; Groll, M.; et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 2012, 31, 1506–1517. [Google Scholar] [CrossRef]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-chaperone Regulation of Conformational Switching in the Hsp90 ATPase Cycle. J. Biol. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef] [PubMed]
- Lott, A.; Oroz, J.; Zweckstetter, M. Molecular basis of the interaction of Hsp90 with its co-chaperone Hop. Protein Sci. 2020, 29, 2422–2432. [Google Scholar] [CrossRef] [PubMed]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric Activation of the Hsp90 Dimer by Its Cochaperone Aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Gohlke, U.; Sobott, F.; Good, V.M.; Ali, M.M.U.; Prodromou, C.; Robinson, C.V.; Saibil, H.R.; Pearl, L.H. Structure of an Hsp90-Cdc37-Cdk4 Complex. Mol. Cell 2006, 23, 697–707. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grindle, M.P.; Carter, B.; Alao, J.P.; Connors, K.; Tehver, R.; Kravats, A.N. Structural Communication between the E. coli Chaperones DnaK and Hsp90. Int. J. Mol. Sci. 2021, 22, 2200. https://doi.org/10.3390/ijms22042200
Grindle MP, Carter B, Alao JP, Connors K, Tehver R, Kravats AN. Structural Communication between the E. coli Chaperones DnaK and Hsp90. International Journal of Molecular Sciences. 2021; 22(4):2200. https://doi.org/10.3390/ijms22042200
Chicago/Turabian StyleGrindle, Matthew P., Ben Carter, John Paul Alao, Katherine Connors, Riina Tehver, and Andrea N. Kravats. 2021. "Structural Communication between the E. coli Chaperones DnaK and Hsp90" International Journal of Molecular Sciences 22, no. 4: 2200. https://doi.org/10.3390/ijms22042200
APA StyleGrindle, M. P., Carter, B., Alao, J. P., Connors, K., Tehver, R., & Kravats, A. N. (2021). Structural Communication between the E. coli Chaperones DnaK and Hsp90. International Journal of Molecular Sciences, 22(4), 2200. https://doi.org/10.3390/ijms22042200