
De Novo Transcriptome Assembly of Two Microsorum Fern Species Identifies Enzymes Required for Two Upstream Pathways of Phytoecdysteroids

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Phenotypes of Two Microsorum Species
2.2. Examination of PE Compounds from Two Microsorum Species
2.3. Library Preparation and RNA-Seq for the Generation of Two Microsorum Transcriptomes
2.4. De Novo Transcriptome Assembly and Taxonomy Classification
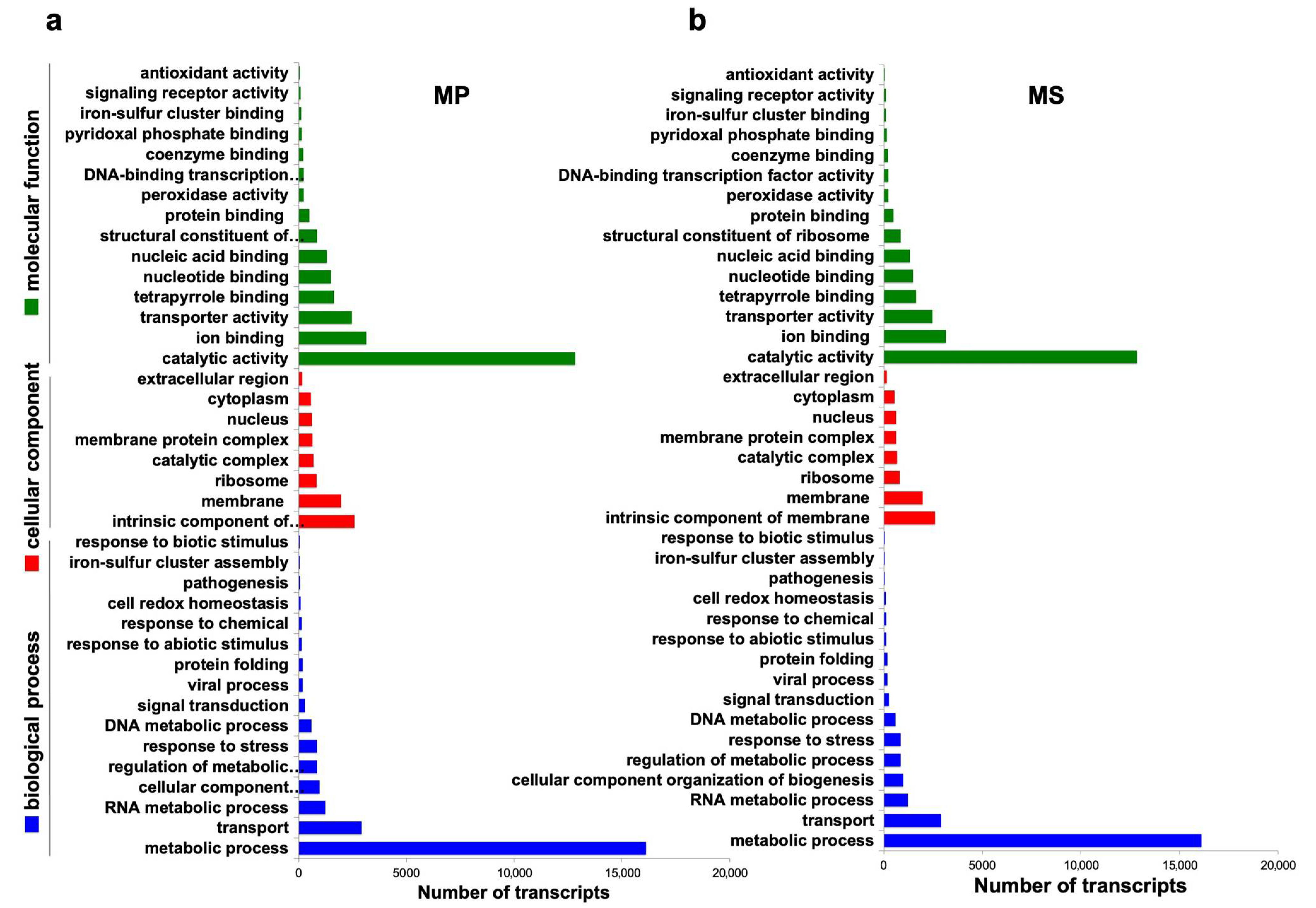
2.5. Functional Annotation of Microsorum Transcripts
2.6. Identification of Microsorum Transcription Factors
2.7. Identification of Putative Microsorum Enzymes Involved in PE Biosynthesis
2.8. Expression Analysis of Genes Involved in the MVA and Sterol-Biosynthetic Pathway
2.9. Phylogenetic Analyses of Enzymes Involved in MVA Pathway
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Measurement of Phytoecdysteroids
4.3. Total RNA Extraction and Transcriptome Sequencing
4.4. De Novo Transcriptome Assembly and Deletion of Contaminated Transcripts
4.5. Annotation of Microsorum Transcripts and Prediction of Open Reading Frames (ORFs)
4.6. Gene-Expression Analysis of the Selected Microsorum Genes
4.7. Real-Time RT-PCR Analysis
4.8. Identification of Transcription Factors and P450 Genes
4.9. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ho, R.; Teai, T.; Girault, J.; Meybeck, A.; Lafont, R.; Raharivelomanana, P. From traditional uses to phytochemical study of a Polynesian healing plant: Constituents and properties of “Metuapua’a”. Planta Medica. 2016, 81, S1–S381. [Google Scholar] [CrossRef]
- Ho, R.; Girault, J.P.; Cousteau, P.Y.; Bianchini, J.P.; Raharivelomanana, P.; Lafont, R. Isolation of a new class of ecdysteroid conjugates (Glucosyl-ferulates) using a combination of liquid chromatographic methods. J. Chromatogr. Sci. 2008, 46, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Teai, T.; Meybeck, A.; Raharivelomanana, P. UV-protective Effects of Phytoecdysteroids from Microsorum grossum Extracts on Human Dermal Fibroblasts. Nat. Prod. Commun. 2015, 10, 33–36. [Google Scholar] [CrossRef]
- Girardi, C.; Butaud, J.F.; Ollier, C.; Ingert, N.; Weniger, B.; Raharivelomanana, P.; Moretti, C. Herbal medicine in the Marquesas Islands. J. Ethnopharmacol. 2015, 161, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Snogan, E.; Vahirua-Lechat, I.; Ho, R.; Bertho, G.; Girault, J.P.; Ortiga, S.; Maria, A.; Lafont, R. Ecdysteroids from the Medicinal Fern Microsorum Scolopendria (Burm. F.). Phytochem. Anal. 2007, 18, 441–450. [Google Scholar] [CrossRef]
- Zibareva, L.; Volodin, V.; Saatov, Z.; Savchenko, T.; Whiting, P.; Lafont, R.; Dinan, L. Distribution of phytoecdysteroids in the Caryophyllaceae. Phytochemistry 2003, 64, 499–517. [Google Scholar] [CrossRef]
- Calas, D.; Thiéry, D.; Marion-Poll, F. 20-Hydroxyecdysone Deters Oviposition and Larval Feeding in the European Grapevine Moth, Lobesia botrana. J. Chem. Ecol. 2006, 32, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.A.; Shaposhnikov, M.V.; Zainullin, V.G.; Punegov, V.V. The influence of ecdysone-containing phyto-preparations on life span of Drosophila melanogaster strains depending on the genotype. Adv. Gerontol. 2006, 19, 33–35. [Google Scholar]
- Voigt, B.; Whiting, P.; Dinan, L. The ecdysteroid agonist/antagonist and brassinosteroid-like activities of synthetic brassinosteroid/ecdysteroid hybrid molecules. Cell. Mol. Life Sci. 2001, 58, 1133–1140. [Google Scholar] [CrossRef]
- Tarkowská, D.; Strnad, M. Isoprenoid-derived plant signaling molecules: Biosynthesis and biological importance. Planta 2018, 247, 1051–1066. [Google Scholar] [CrossRef]
- Soriano, I.R.; Riley, I.T.; Potter, M.J.; Bowers, W.S. Phytoecdysteroids: A novel defense against plant-parasitic nematodes. J. Chem. Ecol. 2004, 30, 1885–1899. [Google Scholar] [CrossRef]
- Jurenka, R.; Russell, K.; O’Neal, M. Phytoecdysteroids as antifeedants towards several beetles that include polyphagous and monophagous feeding guilds. Pest Manag. Sci. 2017, 73, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Gorelick-Feldman, J.; Cohick, W.; Raskin, I. Ecdysteroids elicit a rapid Ca2+ flux leading to Akt activation and increased protein synthesis in skeletal muscle cells. Steroids 2010, 75, 632–637. [Google Scholar] [CrossRef]
- Gorelick-Feldman, J.; MacLean, D.; Ilic, N.; Poulev, A.; Lila, M.A.; Cheng, D.; Raskin, I. Phytoecdysteroids increase protein synthesis in skeletal muscle cells. J. Agric. Food Chem. 2008, 56, 3532–3537. [Google Scholar] [CrossRef]
- Bathori, M.; Pongracz, Z. Phytoecdysteroids from isolation to their effects on humans. Curr. Med. Chem. 2005, 12, 153–172. [Google Scholar] [CrossRef]
- Graf, B.L.; Rojo, L.E.; Delatorre-Herrera, J.; Poulev, A.; Calfio, C.; Raskin, I. Phytoecdysteroids and flavonoid glycosides among chilean and commercial sources of Chenopodium quinoa: Variation and correlation to physico-chemical characteristics. J. Sci. Food Agric. 2016, 96, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Rharrabe, K.; Sayan, F.; Lafont, R. Dietary Effects of Four Phytoecdysteroids on Growth and Development of the Indian Meal Moth, Plodia interpunctella. J. Insect Sci. 2010, 10, 1–12. [Google Scholar] [CrossRef]
- Dinan, L.; Savchenko, T.; Whiting, P. On the distribution of phytoecdysteroids in plants. Cell. Mol. Life Sci. 2001, 58, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Zibareva, L.; Yeriomina, V.I.; Munkhjargal, N.; Girault, J.-P.; Dinan, L.; Lafont, R. The phytoecdysteroid profiles of 7 species of Silene(Caryophyllaceae). Arch. Insect Biochem. Physiol. 2009, 72, 234–248. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Chase, M.W. Trends and concepts in fern classification. Ann. Bot. 2014, 113, 571–594. [Google Scholar] [CrossRef] [PubMed]
- Sessa, E.; Der, J. Evolutionary Genomics of Ferns and Lycophytes. In Advances in Botanical Research; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 215–254. [Google Scholar]
- Li, F.-W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; De Vries, J.; Delaux, P.-M.; Eily, A.; Koppers, N.; Kuo, L.-Y.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.A.; Ponnala, L.; Weber, C.A. Strategies for transcriptome analysis in nonmodel plants. Am. J. Bot. 2012, 99, 267–276. [Google Scholar] [CrossRef]
- Sigel, E.M.; Schuettpelz, E.; Pryer, K.M.; Der, J.P. Overlapping patterns of gene expression between gametophyte and sporophyte phases in the fern Polypodium Amorphum (Polypodiales). Front. Plant Sci. 2018, 9, 1450. [Google Scholar] [CrossRef]
- Zhang, Z.; He, Z.; Xu, S.; Li, X.; Guo, W.; Yang, Y.; Zhong, C.; Zhou, R.; Shi, S. Transcriptome analyses provide insights into the phylogeny and adaptive evolution of the mangrove fern genus Acrostichum. Sci. Rep. 2016, 6, 35634. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hong-Jin, W.; Shu, J.-P.; Zhou, X.-L.; Lei, M.; Wei, R.; Shang, H.; Wei, H.-J.; Zhang, R.; Liu, L.; et al. Large-scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Grusz, A.L.; Rothfels, C.J.; Schuettpelz, E. Transcriptome sequencing reveals genome-wide variation in molecular evolutionary rate among ferns. BMC Genom. 2016, 17, 692. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A Genomic Perspective on Protein Families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.E., Jr.; Churchill, A.C.; Holbrook, N.M. A site for sori: Ecophysiology of fertile-sterile leaf dimorphy in ferns. Am. J. Bot. 2016, 103, 845–855. [Google Scholar] [CrossRef]
- Lee, C.H.; Shin, S.L. Functional Activities of Ferns for Human Health. In Working with Ferns; Springer International Publishing: New York, NY, USA, 2010; pp. 347–359. [Google Scholar]
- Ho, R.; Teai, T.; Bianchini, J.-P.; Lafont, R.; Raharivelomanana, P. Ferns: From Traditional Uses to Pharmaceutical Development, Chemical Identification of Active Principles. In Working with Ferns; Springer International Publishing: New York, NY, USA, 2011; pp. 321–346. [Google Scholar]
- Chen, C.-C.; Liu, H.-Y.; Chen, C.-W.; Schneider, H.; Hyvönen, J. On the spore ornamentation of the microsoroid ferns (microsoroideae, polypodiaceae). J. Plant Res. 2020, 134, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-Y.; Yang, B.; Lan, X.-Y.; Li, X.-Y.; Xu, F.-Y. Cadmium accumulation capacity and resistance strategies of a cadmium-hypertolerant fern—Microsorum fortunei. Sci. Total Environ. 2019, 649, 1209–1223. [Google Scholar] [CrossRef]
- Lan, X.-Y.; Yan, Y.-Y.; Yang, B.; Li, X.-Y.; Xu, F.-L. Differential expression of proteins in the leaves and roots of cadmium-stressed Microsorum pteropus, a novel potential aquatic cadmium hyperaccumulator. Sci. Total. Environ. 2018, 642, 1369–1377. [Google Scholar] [CrossRef]
- Ho, R.; Girault, J.P.; Raharivelomanana, P.; Lafont, R.E. E- and Z-isomers of new phytoecdysteroid conjugates from French Polynesian Microsorum membranifolium (Polypodiaceae) fronds. Molecules 2012, 17, 11598–11606. [Google Scholar] [CrossRef] [PubMed]
- Bosman, M.T. Maria. A monograph of the fern genus Microsorum (Polypodiaceae) including an attempt towards a reconstruction of the phylogenetic history of the Microsoroids. Leiden Bot. Ser. 1991, 14, 1–161. [Google Scholar]
- Jo, Y.; Choi, H.; Cho, J.K.; Yoon, J.-Y.; Choi, S.-K.; Cho, W.K. In silico approach to reveal viral populations in grapevine cultivar Tannat using transcriptome data. Sci. Rep. 2015, 5, 15841. [Google Scholar] [CrossRef] [PubMed]
- Garnica, D.P.; Upadhyaya, N.M.; Dodds, P.N.; Rathjen, J.P. Strategies for wheat stripe rust pathogenicity identified by transcriptome sequencing. PLoS ONE 2013, 8, e67150. [Google Scholar] [CrossRef]
- Kawahara, Y.; Oono, Y.; Kanamori, H.; Matsumoto, T.; Itoh, T.; Minami, E. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS ONE 2012, 7, e49423. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, G.; Pelosi, P. Plant transcriptomes reveal hidden guests. Biochem. Biophys. Res. Commun. 2016, 474, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Lafont, R.; Beydon, P.; Blais, C.; Garcia, M.; Lachaise, F.; Riera, F.; Somme, G.; Girault, J.P. Ecdysteroid metabolism: A comparative study. Ecdysone 1986, 11–16. [Google Scholar] [CrossRef]
- Petryk, A.; Warren, J.T.; Marqués, G.; Jarcho, M.P.; Gilbert, L.I.; Kahler, J.; Parvy, J.P.; Li, Y.; Dauphin-Villemant, C.; O’Connor, M.B. Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. Proc. Natl. Acad. Sci. USA 2003, 100, 13773–13778. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 2016, 34, 441–449. [Google Scholar] [CrossRef]
- Reixach, N.; Irurre-Santillari, J.; Camps, F.; Melé, E.; Messeguer, J.; Casas, J. Phytoecdysteroid overproduction in Polypodium vulgare prothalli. Phytochemistry 1997, 46, 1183–1187. [Google Scholar] [CrossRef]
- Qi, X.; Kuo, L.Y.; Guo, C.; Li, H.; Li, Z.; Qi, J.; Wang, L.; Hu, Y.; Xiang, J.; Zhang, C.; et al. A well-resolved fern nuclear phylogeny reveals the evolution history of numerous transcription factor families. Mol. Phylogenet. Evol. 2018, 127, 961–977. [Google Scholar] [CrossRef]
- Wang, R.; Xu, S.; Wang, N.; Xia, B.; Jiang, Y.; Wang, R. Transcriptome Analysis of Secondary Metabolism Pathway, Transcription Factors, and Transporters in Response to Methyl Jasmonate in Lycoris aurea. Front. Plant Sci. 2017, 7. [Google Scholar] [CrossRef]
- Wang, Q.J.; Li, P.Z.; Sima, Y.H.; Yuan, H.Y.; Wang, J.W. Methyl jasmonate stimulates 20-hydroxyecdysone production in cell suspension cultures of Achyranthes Bidentata. Plant Omics. 2013, 6, 116. [Google Scholar]
- Casimiro-Soriguer, C.S.; Muñoz-Mérida, A.; Pérez-Pulido, A.J. Sma3s: A universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics 2017, 17, 1700071. [Google Scholar] [CrossRef]
- Brunner, A.M.; Yakovlev, I.A.; Strauss, S.H. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Library | Size of Raw Data (bp) | Total Number of Reads | GC Content (%) | Q20 (%) | Accession Number in SRA Database |
|---|---|---|---|---|---|
| MP_R1 | 2,423,192,808 | 23,992,008 | 48.90 | 98.05 | SRR8480531 |
| MP_R2 | 2,275,984,702 | 22,534,502 | 47.87 | 98.17 | SRR8480553 |
| MP_R3 | 2,567,004,688 | 25,415,888 | 47.94 | 98.22 | SRR8480612 |
| MS_R1 | 2,547,634,100 | 25,224,100 | 48.43 | 98.18 | SRR8480631 |
| MS_R2 | 2,340,604,906 | 23,174,306 | 48.36 | 98.22 | SRR8480739 |
| MS_R3 | 2,432,464,810 | 24,083,810 | 48.18 | 98.24 | SRR8480744 |
| Read Statistics | M. punctatum | M. scolopendria | ||
|---|---|---|---|---|
| Preprocessing | Postprocessing | Preprocessing | Postprocessing | |
| Total assembled bases | 111,129,164 | 77,952,463 | 115,715,163 | 90,302,902 |
| Number of transcripts | 131,351 | 57,252 | 106,153 | 54,618 |
| N50 | 1493 | 1934 | 1882 | 2148 |
| Median transcript length | 453 | 1099 | 660 | 1459 |
| Average transcript length | 846 | 1361 | 1090 | 1653 |
| Total number of genes | 80,026 | 27,243 | 64,098 | 23,175 |
| GC content (%) | 45.64 | 45.79 | 45.52 | 45.71 |
| M. punctatum | M. scolopendria | |
|---|---|---|
| Total number of core genes queried | 1440 | 1440 |
| Number of core genes detected (complete) | 952 (66.11%) | 986 (68.47%) |
| Number of core genes detected (complete + partial) | 1023 (71.04%) | 1037 (72.01%) |
| Number of missing core genes | 417 (28.96%) | 403 (27.99%) |
| Average number of orthologs per core genes | 1.77 | 1.99 |
| % of detected core genes that have more than 1 ortholog | 52.73 | 68.97 |
| Pathway | Enzymes | Enzyme no. | No. of Proteins | MP | MS | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MP | MS | Unigene ID | Size | TPM | Unigene ID | Size | TPM | |||
| MVA | Acetoacetyl-CoA transferase (AACT) | 2.3.1.9 | 5 | 3 | MP_TRINITY_ DN32725_c0_g1_i1 | 1254 | 8.97 | MS_TRINITY_ DN30309_c0_g1_i1 | 2001 | 13.79 |
| MP_TRINITY_ DN33165_c0_g2_i2 | 739 | 1.32 | MS_TRINITY_ DN32401_c0_g2_i1 | 1792 | 75.44 | |||||
| MP_TRINITY_ DN34009_c1_g1_i1 | 1774 | 1.53 | MS_TRINITY_ DN36240_c0_g1_i2 | 1523 | 3.06 | |||||
| MP_TRINITY_ DN38258_c0_g1_i2 | 755 | 23.74 | ||||||||
| MP_TRINITY_ DN42159_c0_g1_i1 | 1851 | 21.94 | ||||||||
| 3-Hydroxy-3-methyglutaryl-CoA synthase (HMGS) | 2.3.3.10 | 1 | 1 | MP_TRINITY_ DN41084_c1_g4_i2 | 1751 | 22.11 | MS_TRINITY_ DN32809_c0_g1_i1 | 1969 | 108.60 | |
| 3-Hydroxy-3-methyglutaryl-CoA reductase (HMGR) | 1.1.1.34 | 4 | 2 | MP_TRINITY_ DN38567_c1_g2_i1 | 2322 | 6.24 | MS_TRINITY_ DN12064_c0_g2_i1 | 2498 | 9.16 | |
| MP_TRINITY_ DN40805_c0_g1_i1 | 2429 | 7.49 | MS_TRINITY_ DN35244_c0_g1_i1 | 2708 | 84.46 | |||||
| MP_TRINITY_ DN43604_c0_g1_i1 | 636 | 4.87 | ||||||||
| MP_TRINITY_ DN44231_c0_g4_i1 | 2196 | 245.25 | ||||||||
| Mevalonate kinase (MVK) | 2.7.1.36 | 1 | 1 | MP_TRINITY_ DN35569_c0_g1_i1 | 1503 | 28.41 | MS_TRINITY_ DN31620_c0_g1_i1 | 1622 | 10.26 | |
| Phosphomevalonate kinase (PMK) | 2.7.4.2 | 1 | 1 | MP_TRINITY_ DN40957_c0_g1_i3 | 2108 | 13.19 | MS_TRINITY_ DN31249_c0_g1_i1 | 2212 | 10.26 | |
| Diphospho mevalonate decarboxylase (MVD) | 4.1.1.33 | 2 | 1 | MP_TRINITY_ DN18033_c0_g1_i1 | 471 | 1.09 | MS_TRINITY_ DN32042_c0_g1_i1 | 1604 | 29.62 | |
| MP_TRINITY_ DN38695_c0_g1_i1 | 1710 | 37.22 | ||||||||
| Sterol biosynthesis | isopentenyl diphosphate isomerase (IDI) | 5.3.3.2 | 1 | 1 | MP_TRINITY_ DN40402_c1_g1_i3 | 1297 | 21.93 | MS_TRINITY_ DN30481_c0_g1_i3 | 1244 | 48.19 |
| farnesyl diphosphate synthase (FDS) | 2.5.1.10 | 1 | 2 | MP_TRINITY_ DN38904_c0_g1_i1 | 1662 | 40.55 | MS_TRINITY_ DN30957_c0_g1_i2 | 1382 | 7.46 | |
| MS_TRINITY_ DN31472_c0_g1_i3 | 242 | 63.08 | ||||||||
| squalene synthase (SQS) | 2.5.1.21 | 1 | 2 | MP_TRINITY_ DN38844_c0_g1_i1 | 1521 | 119.78 | MS_TRINITY_ DN31427_c0_g1_i1 | 1456 | 46.17 | |
| MS_TRINITY_ DN45369_c0_g1_i1 | 558 | 1.09 | ||||||||
| Sterol biosyn-thesis | squalene monooxygenase (SQE) | 1.14.13.132 | 2 | 2 | MP_TRINITY_ DN39062_c0_g1_i1 | 2019 | 18.31 | MS_TRINITY_ DN31999_c0_g1_i2 | 1056 | 3.06 |
| MP_TRINITY_ DN43491_c0_g1_i3 | 2273 | 6.95 | MS_TRINITY_ DN33834_c0_g1_i2 | 1926 | 4.83 | |||||
| lanosterol synthase (LAS) | 5.4.99.7 | 3 | 2 | MP_TRINITY_ DN23554_c0_g1_i1 | 680 | 1.44 | MS_TRINITY_ DN248_c0_g1_i1 | 771 | 3.14 | |
| MP_TRINITY_ DN32916_c0_g1_i1 | 560 | 46.28 | MS_TRINITY_ DN31641_c0_g1_i1 | 778 | 32.32 | |||||
| MP_TRINITY_ DN37953_c0_g1_i2 | 2966 | 1.35 | ||||||||
| cycloartenol synthase (CAS) | 5.4.99.8 | 1 | 1 | MP_TRINITY_ DN37919_c0_g1_i1 | 2937 | 24.17 | MS_TRINITY_ DN35135_c0_g1_i1 | 2970 | 12.96 | |
| sterol 14- demethylase (CYP51) | 1.14.13.70 | 3 | 3 | MP_TRINITY_ DN38602_c0_g2_i1 | 1998 | 36.73 | MS_TRINITY_ DN31074_c0_g2_i1 | 2047 | 2.19 | |
| MP_TRINITY_ DN39045_c0_g2_i1 | 1555 | 2.98 | MS_TRINITY_ DN33684_c0_g1_i1 | 1835 | 22.62 | |||||
| MP_TRINITY_ DN39045_c0_g2_i3 | 529 | 1.80 | MS_TRINITY_ DN45802_c0_g1_i1 | 266 | 1.09 | |||||
| δ14-sterol reductase (ERG24) | 1.3.1.70 | 1 | 1 | MP_TRINITY_ DN37114_c0_g1_i1 | 1628 | 6.47 | MS_TRINITY_ DN36333_c2_g1_i1 | 1591 | 4.45 | |
| sterol-4α- carboxylate 3-dehydrogenase (ERG26) | 1.1.1.170 | 2 | 2 | MP_TRINITY_ DN42534_c0_g2_i1 | 1857 | 26.34 | MS_TRINITY_ DN34567_c0_g1_i1 | 1325 | 10.97 | |
| MP_TRINITY_ DN44751_c0_g2_i1 | 1540 | 16.40 | MS_TRINITY_ DN35016_c1_g1_i1 | 2366 | 3.25 | |||||
| δ24-sterol reductase (DHCR24) | 1.3.1.72 | 5 | 2 | MP_TRINITY_ DN31959_c0_g1_i1 | 2111 | 4.07 | MS_TRINITY_ DN28185_c0_g1_i1 | 1932 | 3.69 | |
| MP_TRINITY_ DN32815_c0_g1_i2 | 733 | 2.94 | MS_TRINITY_ DN34498_c0_g1_i1 | 2309 | 13.39 | |||||
| MP_TRINITY_ DN48554_c0_g1_i1 | 210 | 3.11 | ||||||||
| MP_TRINITY_ DN51472_c0_g1_i1 | 258 | 0.45 | ||||||||
| MP_TRINITY_ DN58603_c0_g1_i1 | 220 | 2.04 | ||||||||
| cholestenol δ-isomerase (EBP) | 5.3.3.5 | 1 | 2 | MP_TRINITY_ DN33408_c1_g1_i1 | 973 | 81.36 | MS_TRINITY_ DN24433_c0_g1_i1 | 868 | 5.80 | |
| MS_TRINITY_ DN27664_c0_g1_i1 | 941 | 113.34 | ||||||||
| δ7-sterol 5- desaturase (ERG3) | 1.14.19.20 | 1 | 1 | MP_TRINITY_ DN34658_c0_g1_i1 | 791 | 7.50 | MS_TRINITY_ DN26674_c0_g1_i1 | 1330 | 9.39 | |
| 7-dehydro cholesterol reductase (DHCR7) | 1.3.1.21 | 2 | 2 | MP_TRINITY_ DN37028_c0_g1_i1 | 937 | 22.02 | MS_TRINITY_ DN27356_c0_g1_i1 | 817 | 9.46 | |
| MP_TRINITY_ DN39419_c1_g1_i2 | 2247 | 53.09 | MS_TRINITY_ DN31977_c0_g1_i1 | 2023 | 30.49 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sripinyowanich, S.; Kil, E.-J.; Petchsri, S.; Jo, Y.; Choi, H.; Cho, W.K.; Lee, S. De Novo Transcriptome Assembly of Two Microsorum Fern Species Identifies Enzymes Required for Two Upstream Pathways of Phytoecdysteroids. Int. J. Mol. Sci. 2021, 22, 2085. https://doi.org/10.3390/ijms22042085
Sripinyowanich S, Kil E-J, Petchsri S, Jo Y, Choi H, Cho WK, Lee S. De Novo Transcriptome Assembly of Two Microsorum Fern Species Identifies Enzymes Required for Two Upstream Pathways of Phytoecdysteroids. International Journal of Molecular Sciences. 2021; 22(4):2085. https://doi.org/10.3390/ijms22042085
Chicago/Turabian StyleSripinyowanich, Siriporn, Eui-Joon Kil, Sahanat Petchsri, Yeonhwa Jo, Hoseong Choi, Won Kyong Cho, and Sukchan Lee. 2021. "De Novo Transcriptome Assembly of Two Microsorum Fern Species Identifies Enzymes Required for Two Upstream Pathways of Phytoecdysteroids" International Journal of Molecular Sciences 22, no. 4: 2085. https://doi.org/10.3390/ijms22042085
APA StyleSripinyowanich, S., Kil, E.-J., Petchsri, S., Jo, Y., Choi, H., Cho, W. K., & Lee, S. (2021). De Novo Transcriptome Assembly of Two Microsorum Fern Species Identifies Enzymes Required for Two Upstream Pathways of Phytoecdysteroids. International Journal of Molecular Sciences, 22(4), 2085. https://doi.org/10.3390/ijms22042085