
Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis



{kind=link}
{kind=link}
{kind=link}
Abstract
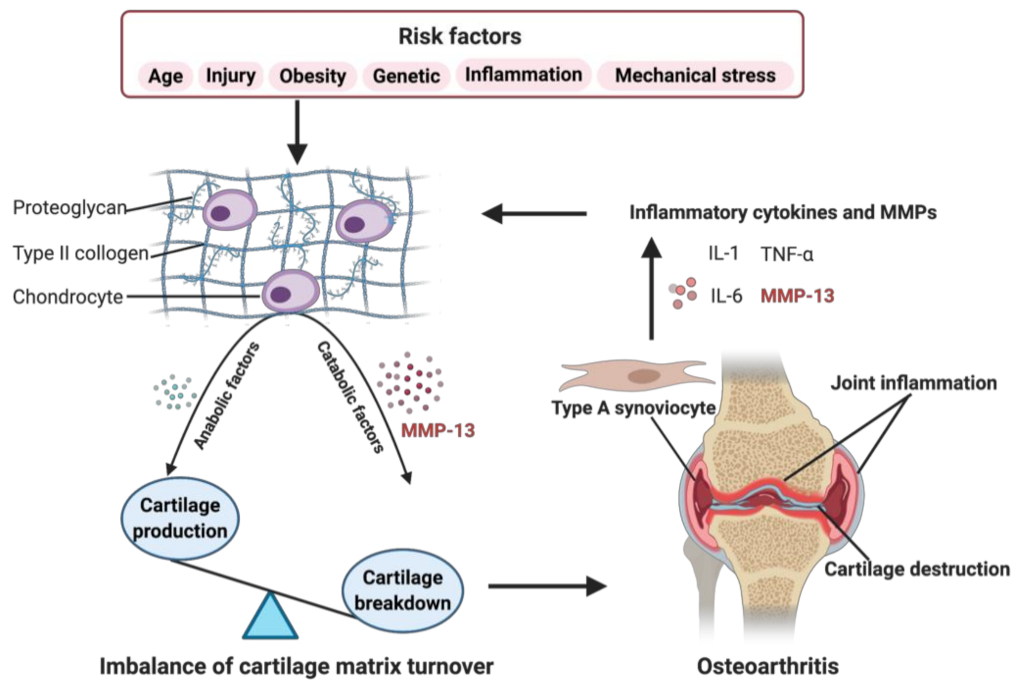
1. Introduction
2. Basic Aspects of MMP-13
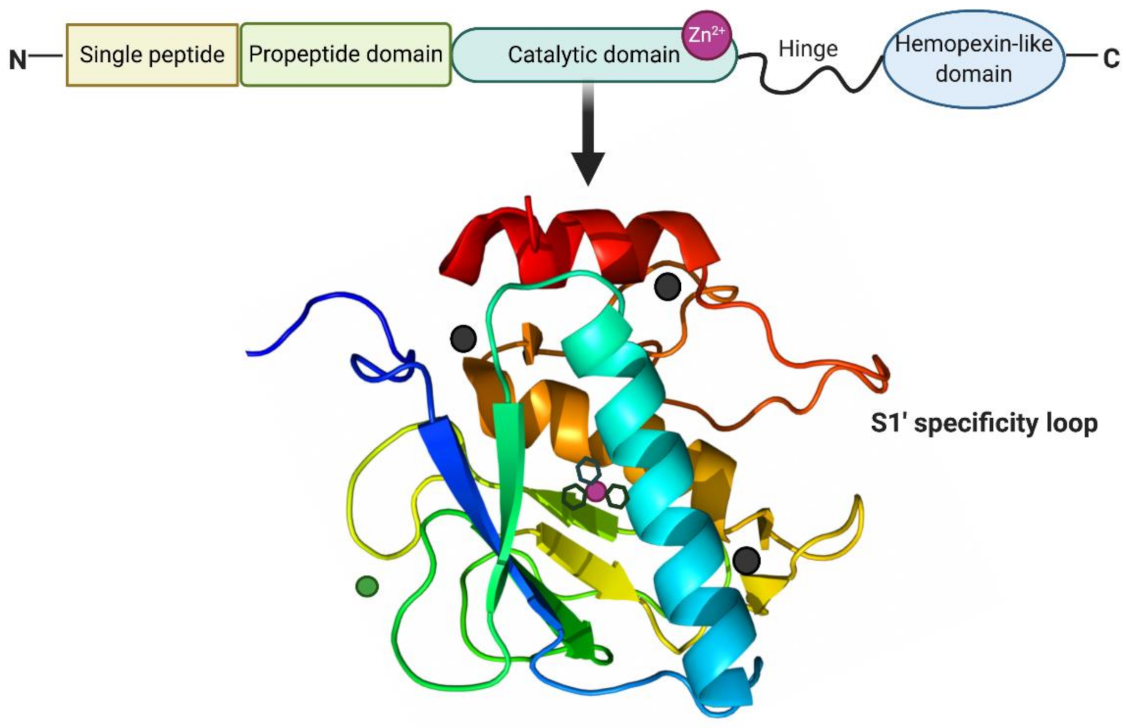
2.1. Structure
2.2. Zymogen Activation
2.3. Role in OA
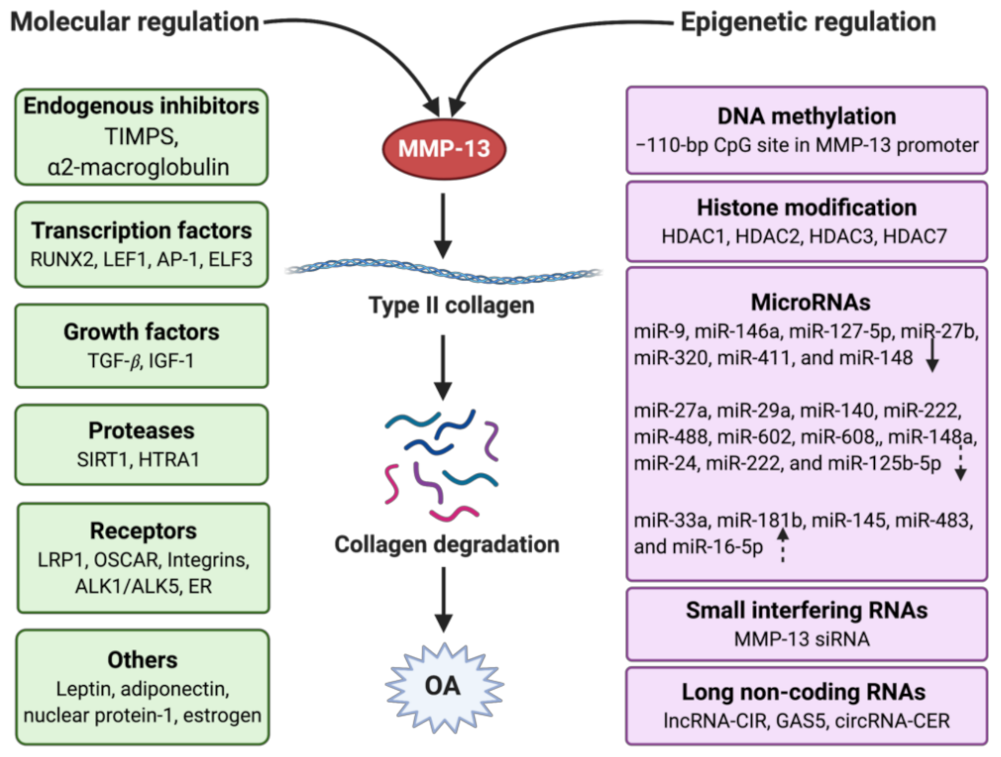
3. Molecular Regulation of MMP-13
3.1. Endogenous Inhibitors
3.2. Transcription Factors
3.3. Growth Factors
3.4. Proteases
3.5. Receptors
3.6. Others
4. Epigenetic Regulation of MMP-13
4.1. DNA Methylation
4.2. Histone Modification
4.3. Non-Coding RNAs
4.3.1. Micro RNA
4.3.2. Small Interfering RNAs (siRNAs)
4.3.3. Long Non-Coding RNAs (lncRNAs)
5. Selective Inhibitors of MMP-13
5.1. Chemically Synthetic Inhibitors
5.1.1. Zinc-Binding Inhibitors
5.1.2. Non-Zinc Binding Inhibitors
5.2. Biologically Synthetic Inhibitor
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Castrogiovanni, P.; Musumeci, G. Which is the best physical treatment for osteoarthritis? J. Funct. Morphol. Kinesiol. 2016, 1, 54–68. [Google Scholar] [CrossRef]
- Litwic, A.; Edwards, M.H.; Dennison, E.M.; Cooper, C. Epidemiology and burden of osteoarthritis. Br. Med. Bull. 2013, 105, 185–199. [Google Scholar] [CrossRef]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018. [Google Scholar] [CrossRef]
- Wilder, F.V.; Hall, B.J.; Barrett, J.P.; Lemrow, N.B. History of acute knee injury and osteoarthritis of the knee: A prospective epidemiological assessment. The clearwater osteoarthritis study. Osteoarthr. Cartil. 2002, 10, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Zhang, Y.Q.; Torner, J.; Nevitt, M.; Lewis, C.E.; Aliabadi, P.; Sack, B.; Clancy, M.; Sharma, L.; Felson, D.T. Is obesity a risk factor for progressive radiographic knee osteoarthritis? Arthritis Care Res. 2009, 61, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, C.; Nguyen, C.; Lefevre-Colau, M.M.; Rannou, F.; Poiraudeau, S. Risk factors and burden of osteoarthritis. Ann. Phys. Rehabil. Med. 2016, 59, 134–138. [Google Scholar] [CrossRef]
- Donell, S. Subchondral bone remodelling in osteoarthritis. EFORT Open Rev. 2019, 4, 221–229. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Little, C.B. The biology of meniscal pathology in osteoarthritis and its contribution to joint disease: Beyond simple mechanics. Connect. Tissue Res. 2017, 58, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; Stocco, E.; Pozzuoli, A.; Granzotto, M.; Porzionato, A.; Vettor, R.; De Caro, R.; Ruggieri, P.; Ramonda, R.; Rossato, M.; et al. Contribution of Infrapatellar Fat Pad and Synovial Membrane to Knee Osteoarthritis Pain. Biomed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Zeng, N.; Yan, Z.P.; Chen, X.Y.; Ni, G.X. Infrapatellar fat pad and knee osteoarthritis. Aging Dis. 2020, 11, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Biologic basis of osteoarthritis: State of the evidence. Curr. Opin. Rheumatol. 2015, 27, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Prim. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint Diseases and Matrix Metalloproteinases: A Role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000, 43, 1916–1926. [Google Scholar] [CrossRef]
- Man, G.S.; Mologhianu, G. Osteoarthritis pathogenesis—A complex process that involves the entire joint. J. Med. Life 2014, 7, 37–41. [Google Scholar] [PubMed]
- Zhang, F.J.; Yu, W.B.; Luo, W.; Gao, S.G.; Li, Y.S.; Lei, G.H. Effect of osteopontin on TIMP-1 and TIMP-2 mRNA in chondrocytes of human knee osteoarthritis in vitro. Exp. Ther. Med. 2014, 8, 391–394. [Google Scholar] [CrossRef]
- Mitchell, P.G.; Magna, H.A.; Reeves, L.M.; Lopresti-Morrow, L.L.; Yocum, S.A.; Rosner, P.J.; Geoghegan, K.F.; Hambor, J.E. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Investing. 1996, 97, 761–768. [Google Scholar] [CrossRef]
- Mueller, M.B.; Tuan, R.S. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: Identifying molecular targets. PM R 2011, 3, S3–S11. [Google Scholar] [CrossRef]
- Di Rosa, M.; Castrogiovanni, P.; Musumeci, G. The synovium theory: Can exercise prevent knee osteoarthritis? The role of “mechanokines”, a possible biological key. J. Funct. Morphol. Kinesiol. 2019, 4, 11. [Google Scholar] [CrossRef]
- Castrogiovanni, P.; Di Rosa, M.; Ravalli, S.; Castorina, A.; Guglielmino, C.; Imbesi, R.; Vecchio, M.; Drago, F.; Szychlinska, M.A.; Musumeci, G. Moderate physical activity as a prevention method for knee osteoarthritis and the role of synoviocytes as biological key. Int. J. Mol. Sci. 2019, 20, 511. [Google Scholar] [CrossRef]
- Chow, Y.Y.; Chin, K.Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediators Inflamm. 2020. [Google Scholar] [CrossRef]
- Sinusas, K. Osteoarthritis: Diagnosis and treatment. Am. Fam. Physician 2012, 85, 49–56. [Google Scholar] [CrossRef]
- Flood, J. The role of acetaminophen in the treatment of osteoarthritis. Am. J. Manag. Care 2010, 16, S48–S54. [Google Scholar] [PubMed]
- Darwiche, H.; Barsoum, W.K.; Klika, A.; Krebs, V.E.; Molloy, R. Retrospective analysis of infection rate after early reoperation in total hip arthroplasty. Proc. Clin. Orthop. Relat. Res. 2010, 468, 2392–2396. [Google Scholar] [CrossRef]
- Mahler, E.A.M.; Minten, M.J.M.; Leseman-Hoogenboom, M.M.; Poortmans, P.M.P.; Leer, J.W.H.; Boks, S.S.; Van Den Hoogen, F.H.J.; Den Broeder, A.A.; Van Den Ende, C.H.M. Effectiveness of low-dose radiation therapy on symptoms in patients with knee osteoarthritis: A randomised, double-blinded, sham-controlled trial. Ann. Rheum. Dis. 2019, 78, 83–90. [Google Scholar] [CrossRef]
- Stevens, R.M.; Ervin, J.; Nezzer, J.; Nieves, Y.; Guedes, K.; Burges, R.; Hanson, P.D.; Campbell, J.N. Randomized, Double-Blind, Placebo-Controlled Trial of Intraarticular Trans-Capsaicin for Pain Associated with Osteoarthritis of the Knee. Arthritis Rheumatol. 2019, 71, 1524–1533. [Google Scholar] [CrossRef]
- Kloppenburg, M.; Peterfy, C.; Haugen, I.K.; Kroon, F.; Chen, S.; Wang, L.; Liu, W.; Levy, G.; Fleischmann, R.M.; Berenbaum, F.; et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1α and anti-interleukin-1β dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 2019, 78, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Smith, C.; Monteith, D.; Brown, R.; Camporeale, A.; McNearney, T.A.; Deeg, M.A.; Raddad, E.; Xiao, N.; de la Peña, A.; et al. CGRP blockade by galcanezumab was not associated with reductions in signs and symptoms of knee osteoarthritis in a randomized clinical trial. Osteoarthr. Cartil. 2018, 26, 1609–1618. [Google Scholar] [CrossRef]
- Jo, C.H.; Lee, Y.G.; Shin, W.H.; Kim, H.; Chai, J.W.; Jeong, E.C.; Kim, J.E.; Shim, H.; Shin, J.S.; Shin, I.S.; et al. Intra-articular injection of mesenchymal stem cells for the treatment of osteoarthritis of the knee: A proof-of-concept clinical trial. Stem Cells 2014, 32, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Meheux, C.J.; McCulloch, P.C.; Lintner, D.M.; Varner, K.E.; Harris, J.D. Efficacy of Intra-articular Platelet-Rich Plasma Injections in Knee Osteoarthritis: A Systematic Review. Arthrosc. J. Arthrosc. Relat. Surg. 2016, 32, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.; Lesur, C.; Thomas, M.; Chomel, A.; Anract, P.; De Nanteuil, G.; Pastoureau, P. Effect of inhibition of matrix metalloproteinases on cartilage loss in vitro and in a guinea pig model of osteoarthritis. Arthritis Rheum. 2005, 52, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742–1749. [Google Scholar] [CrossRef]
- Xie, X.W.; Wan, R.Z.; Liu, Z.P. Recent Research Advances in Selective Matrix Metalloproteinase-13 Inhibitors as Anti-Osteoarthritis Agents. ChemMedChem. 2017, 12, 1157–1168. [Google Scholar] [CrossRef]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J.H. Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2010, 1803, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Sampson, N.S.; Zucker, S.; Cao, J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 2008, 217, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Laronha, H.; Caldeira, J. Structure and Function of Human Matrix Metalloproteinases. Cells 2020, 9, 1076. [Google Scholar] [CrossRef]
- MacColl, E.; Khalil, R.A. Matrix metalloproteinases as regulators of vein structure and function: Implications in chronic venous disease. J. Pharmacol. Exp. Ther. 2015, 355, 410–428. [Google Scholar] [CrossRef]
- Aureli, L.; Gioia, M.; Cerbara, I.; Monaco, S.; Fasciglione, G.; Marini, S.; Ascenzi, P.; Topai, A.; Coletta, M. Structural Bases for Substrate and Inhibitor Recognition by Matrix Metalloproteinases. Curr. Med. Chem. 2008, 15, 2192–2222. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Park, H.I.; Jin, Y.; Hurst, D.R.; Monroe, C.A.; Lee, S.; Schwartz, M.A.; Sang, Q.X.A. The Intermediate S1′ Pocket of the Endometase/Matrilysin-2 Active Site Revealed by Enzyme Inhibition Kinetic Studies, Protein Sequence Analyses, and Homology Modeling. J. Biol. Chem. 2003, 278, 51646–51653. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, B.; Welch, A.R.; Carr, S.; Luong, C.; Broka, C.; Hendricks, R.T.; Campbell, J.A.; Walker, K.A.M.; Martin, R.; Van Wart, H.; et al. Crystal structures of MMP-1 and -13 reveal the structural basis for selectivity of collagenase inhibitors. Nat. Struct. Biol. 1999, 6, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA. 1990, 87, 5578–5582. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Knäuper, V.; Will, H.; López-Otin, C.; Smith, B.; Atkinson, S.J.; Stanton, H.; Hembry, R.M.; Murphy, G. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase A (MMP-2) are able to generate active enzyme. J. Biol. Chem. 1996, 271, 17124–17131. [Google Scholar] [CrossRef]
- Zijlstrat, A.; Aimes, R.T.; Zhu, D.; Regazzoni, K.; Kupriyanova, T.; Seandel, M.; Deryugina, E.I.; Quigley, J.P. Collagenolysis-dependent angiogenesis mediated by matrix metalloproteinase-13 (collagenase-3). J. Biol. Chem. 2004, 279, 27633–27645. [Google Scholar] [CrossRef]
- Rowan, A.D.; Litherland, G.J.; Hui, W.; Milner, J.M. Metalloproteases as potential therapeutic targets in arthritis treatment. Expert Opin. Ther. Targets 2008, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Howes, J.M.; Bihan, D.; Slatter, D.A.; Hamaia, S.W.; Packman, L.C.; Knauper, V.; Visse, R.; Farndale, R.W. The recognition of collagen and triple-helical toolkit peptides by MMP-13: Sequence specificity for binding and cleavage. J. Biol. Chem. 2014, 289, 24091–24101. [Google Scholar] [CrossRef]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.A.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Lemaître, V.; D’Armiento, J.; Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases: Review Article. Pathol. Int. 2010, 60, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar]
- Troeberg, L.; Nagase, H. Analysis of TIMP expression and activity. Methods Mol. Med. 2007, 135, 251–267. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Sahebjam, S.; Khokha, R.; Mort, J.S. Increased collagen and aggrecan degradation with age in the joints of Timp3-/-mice. Arthritis Rheum. 2007, 56, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Castner, B.; Slack, J.; Tocker, J.; Eisenman, J.; Jacobson, E.; Delaney, J.; Winters, D.; Hecht, R.; Bendele, A. A14 Injected Timp-3 protects cartilage in a rat meniscal tear model. Osteoarthr. Cartil. 2006, 14, S23–S24. [Google Scholar] [CrossRef][Green Version]
- Kevorkian, L.; Young, D.A.; Darrah, C.; Donell, S.T.; Shepstone, L.; Porter, S.; Brockbank, S.M.V.; Edwards, D.R.; Parker, A.E.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50, 131–141. [Google Scholar] [CrossRef]
- Strickland, D.K.; Ashcom, J.D.; Williams, S.; Burgess, W.H.; Migliorini, M.; Scott Argraves, W. Sequence identity between the α2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J. Biol. Chem. 1990, 265, 17401–17404. [Google Scholar] [CrossRef]
- Chen, D.; Kim, D.J.; Shen, J.; Zou, Z.; O’Keefe, R.J. Runx2 plays a central role in Osteoarthritis development. J. Orthop. Transl. 2020, 23, 132–139. [Google Scholar] [CrossRef]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Saito, T.; Yano, F.; Ikeda, T.; Mabuchi, A.; Sapkota, B.R.; Akune, T.; Nishida, N.; et al. C/EBPβ and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2α as the inducer in chondrocytes. Hum. Mol. Genet. 2012, 21, 1111–1123. [Google Scholar] [CrossRef]
- Yun, K.; Im, S.H. Transcriptional regulation of MMP13 by Lef1 in chondrocytes. Biochem. Biophys. Res. Commun. 2007, 364, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.; So, J.-S.; Jash, A.; Im, S.-H. Lymphoid Enhancer Binding Factor 1 Regulates Transcription through Gene Looping. J. Immunol. 2009, 183, 5129–5137. [Google Scholar] [CrossRef] [PubMed]
- Grall, F.; Gu, X.; Tan, L.; Cho, J.Y.; Inan, M.S.; Pettit, A.R.; Thamrongsak, U.; Choy, B.K.; Manning, C.; Akbarali, Y.; et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor α in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor κB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003, 48, 1249–1260. [Google Scholar] [CrossRef]
- Otero, M.; Plumb, D.A.; Tsuchimochi, K.; Dragomir, C.L.; Hashimoto, K.; Peng, H.; Olivotto, E.; Bevilacqua, M.; Tan, L.; Yang, Z.; et al. E74-like Factor 3 (ELF3) impacts on Matrix Metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem. 2012, 287, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Hosaka, Y.; Okada, K.; Mori, D.; Yano, F.; Kobayashi, H.; Taniguchi, Y.; Mori, Y.; Okuma, T.; Chang, S.H.; et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA. 2015, 112, 3080–3085. [Google Scholar] [CrossRef] [PubMed]
- Pombo-Suarez, M.; Castaño-Oreja, M.T.; Calaza, M.; Gomez-Reino, J.; Gonzalez, A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 2009, 68, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Liu, M.; Harper, P.E.; Doré, J.; Martin, G.; Furey, A.; Green, R.; Rahman, P.; Zhai, G. Overexpression of MMP13 in human osteoarthritic cartilage is associated with the SMAD-independent TGF-β signalling pathway. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Massicotte, F.; Aubry, I.; Martel-Pelletier, J.; Pelletier, J.P.; Fernandes, J.; Lajeunesse, D. Abnormal insulin-like growth factor 1 signaling in human osteoarthritic subchondral bone osteoblasts. Arthritis Res. Ther. 2006, 8. [Google Scholar] [CrossRef]
- Zhang, M.; Zhou, Q.; Liang, Q.Q.; Li, C.G.; Holz, J.D.; Tang, D.; Sheu, T.J.; Li, T.F.; Shi, Q.; Wang, Y.J. IGF-1 regulation of type II collagen and MMP-13 expression in rat endplate chondrocytes via distinct signaling pathways. Osteoarthr. Cartil. 2009, 17, 100–106. [Google Scholar] [CrossRef]
- Dvir-Ginzberg, M.; Gagarina, V.; Lee, E.J.; Booth, R.; Gabay, O.; Hall, D.J. Tumor necrosis factor α-mediated cleavage and inactivation of sirT1 in human osteoarthritic chondrocytes. Arthritis Rheum. 2011, 63, 2363–2373. [Google Scholar] [CrossRef]
- Elayyan, J.; Lee, E.J.; Gabay, O.; Smith, C.A.; Qiq, O.; Reich, E.; Mobasheri, A.; Henrotin, Y.; Kimber, S.J.; Dvir-Ginzberg, M. LEF1-mediated MMP13 gene expression is repressed by SIRT1 in human chondrocytes. FASEB J. 2017, 31, 3116–3125. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, H.; Hu, B.; Zhang, M. Sirt1 regulates apoptosis and extracellular matrix degradation in resveratrol-treated osteoarthritis chondrocytes via the wnt/β-catenin signaling pathways. Exp. Ther. Med. 2017, 14, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Polur, I.; Lee, P.L.; Servais, J.M.; Xu, L.; Li, Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol. Histopathol. 2010, 25, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Servais, J.; Polur, I.; Kim, D.; Lee, P.L.; Chung, K.; Li, Y. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010, 62, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Okano, H.; Miyagawa, W.; Visse, R.; Shitomi, Y.; Santamaria, S.; Dudhia, J.; Troeberg, L.; Strickland, D.K.; Hirohata, S.; et al. MMP-13 is constitutively produced in human chondrocytes and co-endocytosed with ADAMTS-5 and TIMP-3 by the endocytic receptor LRP1. Matrix Biol. 2016, 56, 57–73. [Google Scholar] [CrossRef]
- Park, D.R.; Kim, J.; Kim, G.M.; Lee, H.; Kim, M.; Hwang, D.; Lee, H.; Kim, H.S.; Kim, W.; Park, M.C.; et al. Osteoclast-associated receptor blockade prevents articular cartilage destruction via chondrocyte apoptosis regulation. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F. Integrins and chondrocyte-matrix interactions in articular cartilage. Matrix Biol. 2014, 39, 11–16. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Pulai, J.; Loeser, R.F. Fibronectin fragments and blocking antibodies to α2β1 and α5β1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002, 46, 2368–2376. [Google Scholar] [CrossRef]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.G.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.-J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 Ratio as a Cause for Elevated MMP-13 Expression in Osteoarthritis in Humans and Mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Xu, Z.; Yin, Y.; Su, J.; Niu, X.; Cao, X. Effects of estradiol on reduction of osteoarthritis in rabbits through effect on matrix metalloproteinase proteins. Iran. J. Basic Med. Sci. 2016, 19, 310–315. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Malizos, K.N.; Tsezou, A. Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: Possible molecular target for osteoarthritis therapeutic intervention. Ann. Rheum. Dis. 2007, 66, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Francin, P.J.; Abot, A.; Guillaume, C.; Moulin, D.; Bianchi, A.; Gegout-Pottie, P.; Jouzeau, J.Y.; Mainard, D.; Presle, N. Association between adiponectin and cartilage degradation in human osteoarthritis. Osteoarthr. Cartil. 2014, 22, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Yammani, R.R.; Loeser, R.F. Brief report: Stress-inducible nuclear protein 1 regulates matrix metalloproteinase 13 expression in human articular chondrocytes. Arthritis Rheumatol. 2014, 66, 1266–1271. [Google Scholar] [CrossRef]
- Liang, Y.; Duan, L.; Xiong, J.; Zhu, W.; Liu, Q.; Wang, D.; Liu, W.; Li, Z.; Wang, D. E2 regulates MMP-13 via targeting miR-140 in IL-1β-induced extracellular matrix degradation in human chondrocytes. Arthritis Res. Ther. 2016, 18. [Google Scholar] [CrossRef]
- Romanoski, C.E.; Glass, C.K.; Stunnenberg, H.G.; Wilson, L.; Almouzni, G. Epigenomics: Roadmap for regulation. Nature 2015, 518, 314–316. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Reynard, L.N.; Loughlin, J. Genetics and epigenetics of osteoarthritis. Maturitas 2012, 71, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Otero, M.; Imagawa, K.; De Andrés, M.C.; Coico, J.M.; Roach, H.I.; Oreffo, R.O.C.; Marcu, K.B.; Goldring, M.B. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J. Biol. Chem. 2013, 288, 10061–10072. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Yamada, N.; Cheung, K.S.C.; Tilley, S.; Clarke, N.M.P.; Oreffo, R.O.C.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef]
- Cheung, K.S.C.; Hashimoto, K.; Yamada, N.; Roach, H.I. Expression of ADAMTS-4 by chondrocytes in the surface zone of human osteoarthritic cartilage is regulated by epigenetic DNA de-methylation. Rheumatol. Int. 2009, 29, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Goldring, M.B.; Oreffo, R.O.C. DNA methylation of the RUNX2 P1 promoter mediates MMP13 transcription in chondrocytes. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Khan, N.M.; Haqqi, T.M. Epigenetics in osteoarthritis: Potential of HDAC inhibitors as therapeutics. Pharmacol. Res. 2018, 128, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced Histone Acetylation and Transcription: A Dynamic Perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef]
- Hong, S.; Derfoul, A.; Pereira-Mouries, L.; Hall, D.J. A novel domain in histone deacetylase 1 and 2 mediates repression of cartilage-specific genes in human chondrocytes. FASEB J. 2009, 23, 3539–3552. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, R.; Miyaki, S.; Yamashita, S.; Yoshitaka, T.; Lindman, G.; Ito, Y.; Sasho, T.; Takahashi, K.; Lotz, M.; Asahara, H. Correlation between MMP-13 and HDAC7 expression in human knee osteoarthritis. Mod. Rheumatol. 2010, 20, 11–17. [Google Scholar] [CrossRef]
- Carpio, L.R.; Bradley, E.W.; Westendorf, J.J. Histone deacetylase 3 suppresses Erk phosphorylation and matrix metalloproteinase (Mmp)-13 activity in chondrocytes. Connect. Tissue Res. 2017, 58, 27–36. [Google Scholar] [CrossRef]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Barter, M.J.; Young, D.A. Epigenetic mechanisms and non-coding rnas in osteoarthritis. Curr. Rheumatol. Rep. 2013, 15. [Google Scholar] [CrossRef]
- Sondag, G.R.; Haqqi, T.M. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Eulalio, A.; Huntzinger, E.; Nishihara, T.; Rehwinkel, J.; Fauser, M.; Izaurralde, E. Deadenylation is a widespread effect of miRNA regulation. RNA 2009, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Asahara, H. Current Status and Strategy of microRNA Research for Cartilage Development and Osteoarthritis Pathogenesis. J. Bone Metab. 2016, 23, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Song, B.; Pan, Z. Downregulation of microRNA-9 increases Matrix metalloproteinase-13 expression levels and facilitates osteoarthritis onset. Mol. Med. Rep. 2018, 17, 3708–3714. [Google Scholar] [CrossRef]
- Gu, R.; Liu, N.; Luo, S.; Huang, W.; Zha, Z.; Yang, J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine 2016, 95, e4315. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Chun, C.H.; Jin, E.J. MicroRNA-9 regulates survival of chondroblasts and cartilage integrity by targeting protogenin. Cell Commun. Signal. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of microRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009, 60, 1035–1041. [Google Scholar] [CrossRef]
- Meng, F.; Zhang, Z.; Chen, W.; Huang, G.; He, A.; Hou, C.; Long, Y.; Yang, Z.; Zhang, Z.; Liao, W. MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1β-induced chondrocyte responses. Osteoarthr. Cartil. 2016, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. MicroRNA-127-5p regulates matrix metalloproteinase 13 expression and interleukin-1β-induced catabolic effects in human chondrocytes. Arthritis Rheum. 2013, 65, 3141–3152. [Google Scholar] [CrossRef]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Y.; Zhao, X.; Meng, C.; Ma, L.; Kong, Y. MicroRNA-411 inhibited matrix metalloproteinase 13 expression in human chondrocytes. Am. J. Transl. Res. 2015, 7, 2000–2006. [Google Scholar]
- Vonk, L.A.; Kragten, A.H.M.; Dhert, W.J.A.; Saris, D.B.F.; Creemers, L.B. Overexpression of hsa-miR-148a promotes cartilage production and inhibits cartilage degradation by osteoarthritic chondrocytes. Osteoarthr. Cartil. 2014, 22, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Hum, D.; Pelletier, J.P.; Duval, N.; Martel-Pelletier, J. Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskelet. Disord. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhen, Z.; Tang, G.; Zheng, C.; Yang, G. MiR-29a and MiR-140 protect chondrocytes against the anti-proliferation and cell matrix signaling changes by IL-1β. Mol. Cells 2016, 39, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jin, E.H.; Kim, D.; Kim, K.Y.; Chun, C.H.; Jin, E.J. MicroRNA-222 regulates MMP-13 via targeting HDAC-4 during osteoarthritis pathogenesis. BBA Clin. 2015, 3, 79–89. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Lee, C.H.; Lee, M.S.; Chun, C.H.; Jin, E.J. MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during pathogenesis of osteoarthritis. J. Biomed. Sci. 2013, 20. [Google Scholar] [CrossRef]
- Akhtar, N.; Makki, M.S.; Haqqi, T.M. MicroRNA-602 and microRNA-608 regulate sonic hedgehog expression via target sites in the coding region in human chondrocytes. Arthritis Rheumatol. 2015, 67, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Philipot, D.; Guérit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. P16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Rasheed, N.; Abdulmonem, W.A.; Khan, M.I. MicroRNA-125b-5p regulates IL-1β induced inflammatory genes via targeting TRAF6-mediated MAPKs and NF-κB signaling in human osteoarthritic chondrocytes. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, F.; Malizos, K.N.; Papathanasiou, I.; Tsezou, A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef]
- Song, J.; Lee, M.; Kim, D.; Han, J.; Chun, C.H.; Jin, E.J. MicroRNA-181b regulates articular chondrocytes differentiation and cartilage integrity. Biochem. Biophys. Res. Commun. 2013, 431, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kang, X.; Xing, Y.; Dou, C.; Kang, F.; Li, J.; Quan, Y.; Dong, S. Effect of microRNA-145 on IL-1β-induced cartilage degradation in human chondrocytes. FEBS Lett. 2014, 588, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, J.; Liu, X.; Yang, S.; Ye, S.; Yang, W.; Zhang, Y. MicroRNA-16-5p Controls Development of Osteoarthritis by Targeting SMAD3 in Chondrocytes. Curr. Pharm. Des. 2015, 21, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ma, N.; Yan, F.; Yu, Z.; Wu, G.; Qiao, Y.; Han, D.; Xiang, Y.; Li, F.; Wang, W.; et al. The expression of intronic miRNAs, miR-483 and miR-483*, and their host gene, Igf2, in murine osteoarthritis cartilage. Int. J. Biol. Macromol. 2013, 61, 43–49. [Google Scholar] [CrossRef]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, H.; Akagi, R.; Yamaguchi, S.; Muramatsu, Y.; Akatsu, Y.; Yamamoto, Y.; Sasaki, T.; Takahashi, K.; Sasho, T. Effect of inhibiting MMP13 and ADAMTS5 by intra-articular injection of small interfering RNA in a surgically induced osteoarthritis model of mice. Cell Tissue Res. 2017, 368, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Akagi, R.; Sasho, T.; Saito, M.; Endo, J.; Yamaguchi, S.; Muramatsu, Y.; Mukoyama, S.; Akatsu, Y.; Katsuragi, J.; Fukawa, T.; et al. Effective knock down of matrix metalloproteinase-13 by an intra-articular injection of small interfering RNA (siRNA) in a murine surgically-induced osteoarthritis model. J. Orthop. Res. 2014, 32, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, H.; Sasho, T.; Akagi, R.; Muramatsu, Y.; Mukoyama, S.; Akatsu, Y.; Fukawa, T.; Katsuragi, J.; Endo, J.; Yamamoto, Y. Effective knock down of MMP13 and ADAMTS5 by intra-articular injection of small interference RNA (siRNA) in a surgically induced osteoarthritis model of mice. Osteoarthr. Cartil. 2014, 22, S372–S373. [Google Scholar] [CrossRef][Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Huang, G.; Zhang, Z.; Liu, J.; Zhang, Z.; Huang, Z.; Yu, B.; Meng, F. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthr. Cartil. 2015, 23, 423–432. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Dai, L.; Hu, X.; Zhu, J.; Li, L.; Zhou, C.; Ao, Y. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol. 2014, 66, 969–978. [Google Scholar] [CrossRef]
- Song, J.; Ahn, C.; Chun, C.H.; Jin, E.J. A long non-coding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J. Orthop. Res. 2014, 32, 1628–1635. [Google Scholar] [CrossRef]
- Wang, G.; Bu, X.; Zhang, Y.; Zhao, X.; Kong, Y.; Ma, L.; Niu, S.; Wu, B.; Meng, C. LncRNA-UCA1 enhances MMP-13 expression by inhibiting miR- 204-5p in human chondrocytes. Oncotarget 2017, 8, 91281–91290. [Google Scholar] [CrossRef]
- Seda Yar Saglam, A.; Alp, E.; Ilke Onen, H. Circular RNAs and Its Biological Functions in Health and Disease. In Gene Expression and Phenotypic Traits; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Hu, X.; Dai, L.; Fu, X.; Zhang, J.; Ao, Y. Circular RNA Related to the Chondrocyte ECM Regulates MMP13 Expression by Functioning as a MiR-136 “Sponge” in Human Cartilage Degradation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Gu, H.; Jiao, Y.; Yu, X.; Li, X.; Wang, W.; Ding, L.; Liu, L. Resveratrol inhibits the IL-1β-induced expression ofMMP-13 and IL-6 in human articular chondrocytes viaTLR4/MyD88-dependent and-independent signaling cascades. Int. J. Mol. Med. 2017, 39, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wu, S.; Mao, X.; Wang, W.; Tai, H. Inhibition effect of curcumin on TNF-α and MMP-13 expression induced by advanced glycation end products in chondrocytes. Pharmacology 2013, 91, 77–85. [Google Scholar] [CrossRef]
- Rasheed, Z.; Anbazhagan, A.N.; Akhtar, N.; Ramamurthy, S.; Voss, F.R.; Haqqi, T.M. Green tea polyphenol epigallocatechin-3-gallate inhibits advanced glycation end product-induced expression of tumor necrosis factor-α and matrix metalloproteinase-13 in human chondrocytes. Arthritis Res. Ther. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Janusz, M.J.; Hookfin, E.B.; Heitmeyer, S.A.; Woessner, J.F.; Freemont, A.J.; Hoyland, J.A.; Brown, K.K.; Hsieh, L.C.; Almstead, N.G.; De, B.; et al. Moderation of iodoacetate-induced experimental osteoarthritis in rats by matrix metalloproteinase inhibitors. Osteoarthr. Cartil. 2001, 9, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix Metalloproteinases as Valid Clinical Target. Curr. Pharm. Des. 2006, 13, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Pavlovsky, A.G.; Ortwine, D.F.; Prior, F.; Man, C.F.; Bornemeier, D.A.; Banotai, C.A.; Mueller, W.T.; McConnell, P.; Yan, C.; et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 2007, 282, 27781–27791. [Google Scholar] [CrossRef]
- Nuti, E.; Casalini, F.; Avramova, S.I.; Santamaria, S.; Cercignani, G.; Marinelli, L.; La Pietra, V.; Novellino, E.; Orlandini, E.; Nencetti, S.; et al. N-O-isopropyl sulfonamido-based hydroxamates: Design, synthesis and biological evaluation of selective matrix metalloproteinase-13 inhibitors as potential therapeutic agents for osteoarthritis. J. Med. Chem. 2009, 52, 4757–4773. [Google Scholar] [CrossRef]
- Monovich, L.G.; Tommasi, R.A.; Fujimoto, R.A.; Blancuzzi, V.; Clark, K.; Cornell, W.D.; Doti, R.; Doughty, J.; Fang, J.; Farley, D.; et al. Discovery of potent, selective, and orally active carboxylic acid based inhibitors of matrix metalloproteinase-13. J. Med. Chem. 2009, 52, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- Nara, H.; Sato, K.; Kaieda, A.; Oki, H.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; Uchikawa, O.; Kori, M. Design, synthesis, and biological activity of novel, potent, and highly selective fused pyrimidine-2-carboxamide-4-one-based matrix metalloproteinase (MMP)-13 zinc-binding inhibitors. Bioorg. Med. Chem. 2016, 24, 6149–6165. [Google Scholar] [CrossRef]
- Nara, H.; Kaieda, A.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; et al. Discovery of novel, highly potent, and selective matrix metalloproteinase (MMP)-13 inhibitors with a 1,2,4-triazol-3-yl moiety as a zinc binding group using a structure-based design approach. J. Med. Chem. 2017, 60, 608–626. [Google Scholar] [CrossRef] [PubMed]
- De Savi, C.; Morley, A.D.; Ting, A.; Nash, I.; Karabelas, K.; Wood, C.M.; James, M.; Norris, S.J.; Karoutchi, G.; Rankine, N.; et al. Selective non zinc binding inhibitors of MMP13. Bioorg. Med. Chem. Lett. 2011, 21, 4215–4219. [Google Scholar] [CrossRef]
- Jie, J.L.; Nahra, J.; Johnson, A.R.; Bunker, A.; O’Brien, P.; Yue, W.S.; Ortwine, D.F.; Man, C.F.; Baragi, V.; Kilgore, K.; et al. Quinazolinones and pyrido[3,4-d]pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2008, 51, 835–841. [Google Scholar] [CrossRef]
- Schnute, M.E.; O’Brien, P.M.; Nahra, J.; Morris, M.; Howard Roark, W.; Hanau, C.E.; Ruminski, P.G.; Scholten, J.A.; Fletcher, T.R.; Hamper, B.C.; et al. Discovery of (pyridin-4-yl)-2H-tetrazole as a novel scaffold to identify highly selective matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. Bioorg. Med. Chem. Lett. 2010, 20, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Gege, C.; Bao, B.; Bluhm, H.; Boer, J.; Gallagher, B.M.; Korniski, B.; Powers, T.S.; Steeneck, C.; Taveras, A.G.; Baragi, V.M. Discovery and evaluation of a non-Zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J. Med. Chem. 2012, 55, 709–716. [Google Scholar] [CrossRef]
- Settle, S.; Vickery, L.; Nemirovskiy, O.; Vidmar, T.; Bendele, A.; Messing, D.; Ruminski, P.; Schnute, M.; Sunyer, T. Cartilage degradation biomarkers predict efficacy of a novel, highly selective matrix metalloproteinase 13 inhibitor in a dog model of osteoarthritis: Confirmation by multivariate analysis that modulation of type II collagen and aggrecan degradation pepti. Arthritis Rheum. 2010, 62, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Ruminski, P.G.; Massa, M.; Strohbach, J.; Hanau, C.E.; Schmidt, M.; Scholten, J.A.; Fletcher, T.R.; Hamper, B.C.; Carroll, J.N.; Shieh, H.S.; et al. Discovery of N-(4-Fluoro-3-methoxybenzyl)-6-(2-(((2S,5R)-5-(hydroxymethyl)-1,4-dioxan-2-yl)methyl)-2H-tetrazol-5-yl)-2-methylpyrimidine-4-carboxamide. A Highly Selective and Orally Bioavailable Matrix Metalloproteinase-13 Inhibitor for the Potential Treat. J. Med. Chem. 2016, 59, 313–327. [Google Scholar] [CrossRef]
- Zheng, S.; Hunter, D.J.; Xu, J.; Ding, C. Monoclonal antibodies for the treatment of osteoarthritis. Expert Opin. Biol. Ther. 2016, 16, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; Takahashi, T.; Onoda, J.; Yamauchi, A.; Kawai, T.; Kishino, J.; Yamane, S.; Fujii, I.; Fukui, N.; Numata, Y. Development of a neutralizing antibody specific for the active form of matrix metalloproteinase-13. Biochemistry 2012, 51, 8877–8884. [Google Scholar] [CrossRef]
- Amar, S.; Fields, G.B. Potential clinical implications of recent matrix metalloproteinase inhibitor design strategies. Expert Rev. Proteomics 2015, 12, 445–447. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Q.; Ecker, M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 1742. https://doi.org/10.3390/ijms22041742
Hu Q, Ecker M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. International Journal of Molecular Sciences. 2021; 22(4):1742. https://doi.org/10.3390/ijms22041742
Chicago/Turabian StyleHu, Qichan, and Melanie Ecker. 2021. "Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis" International Journal of Molecular Sciences 22, no. 4: 1742. https://doi.org/10.3390/ijms22041742
APA StyleHu, Q., & Ecker, M. (2021). Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. International Journal of Molecular Sciences, 22(4), 1742. https://doi.org/10.3390/ijms22041742