
Paroxetine—Overview of the Molecular Mechanisms of Action




Abstract
1. Introduction
2. Paroxetine—General Information and History
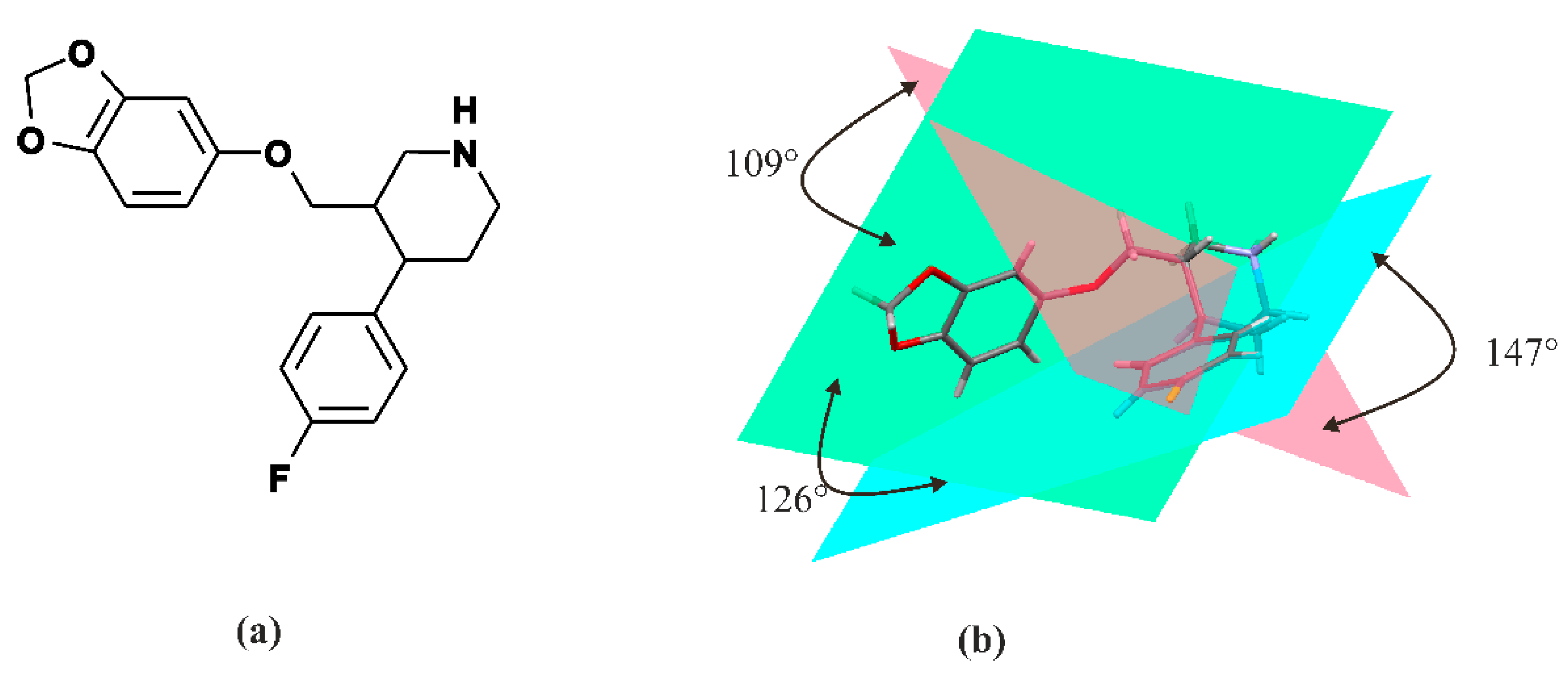
3. Paroxetine—Chemistry
4. Paroxetine—Pharmacodynamics Pharmacokinetics and Metabolism
4.1. Paroxetine as P450 Inhibitors
4.1.1. Paroxetine as CYP2D6 Inhibitors
4.1.2. Paroxetine as CYP2B6 Inhibitors
4.1.3. Paroxetine as CYP2B4 Inhibitors
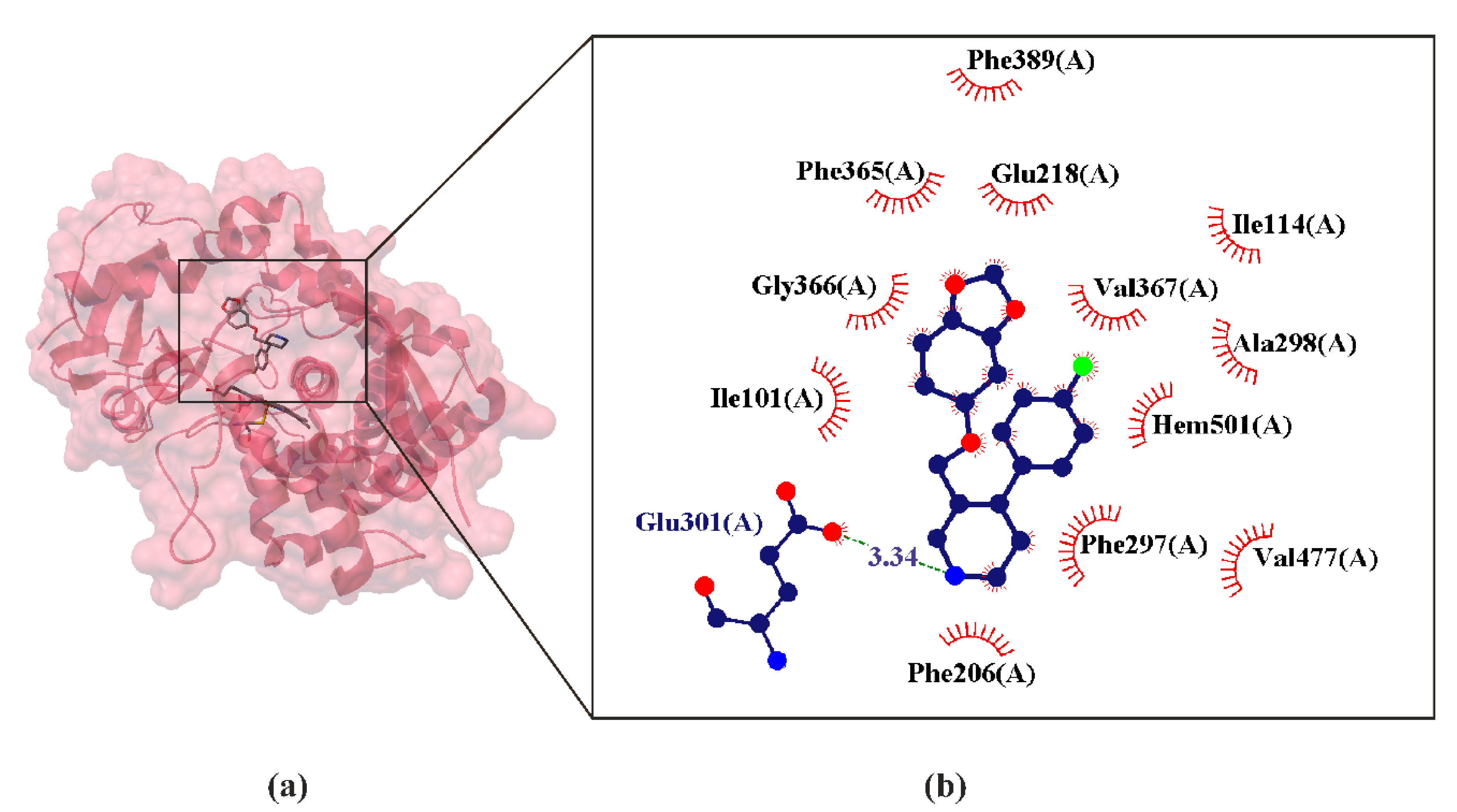
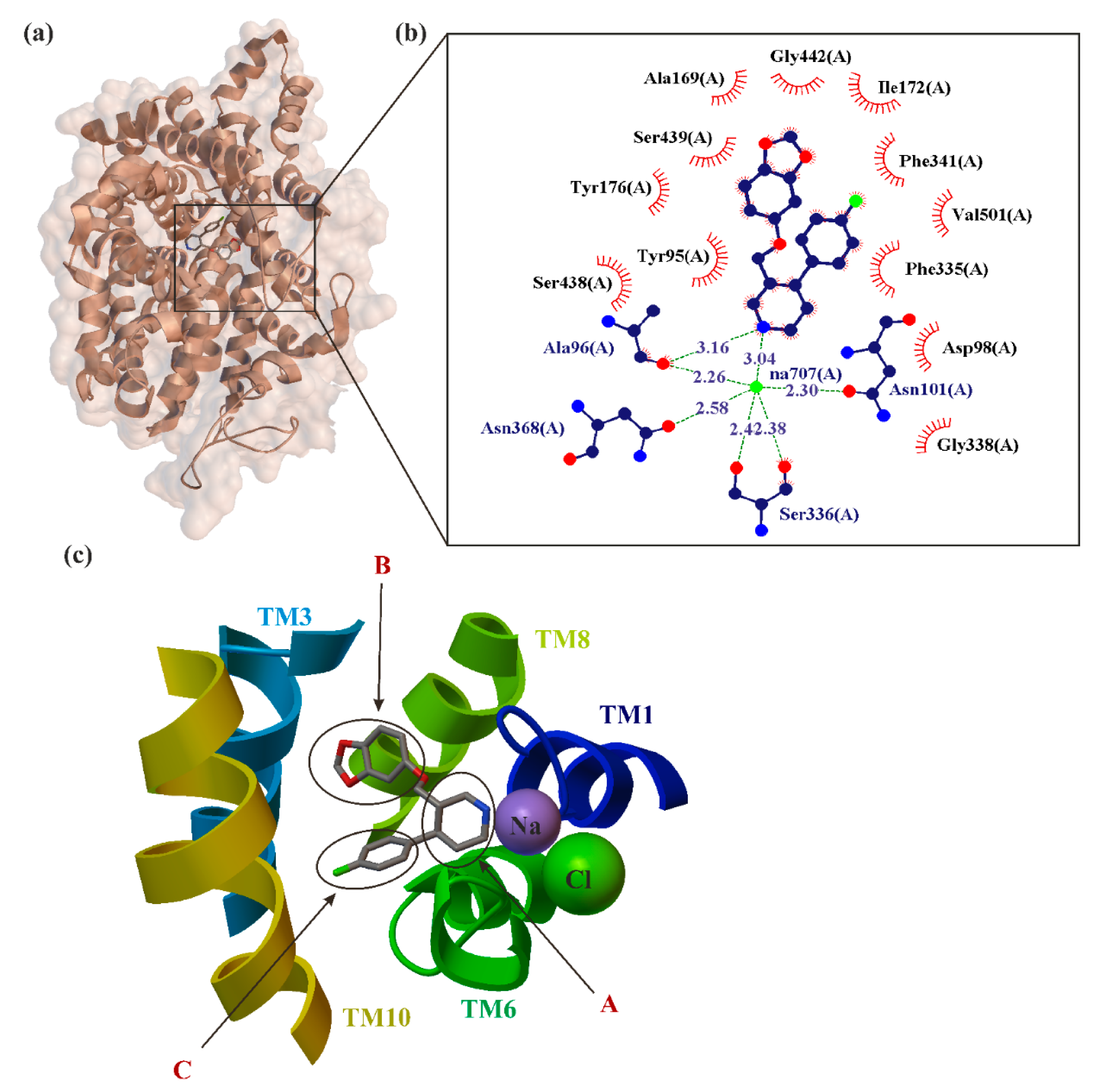
5. Paroxetine as MAT Inhibitions
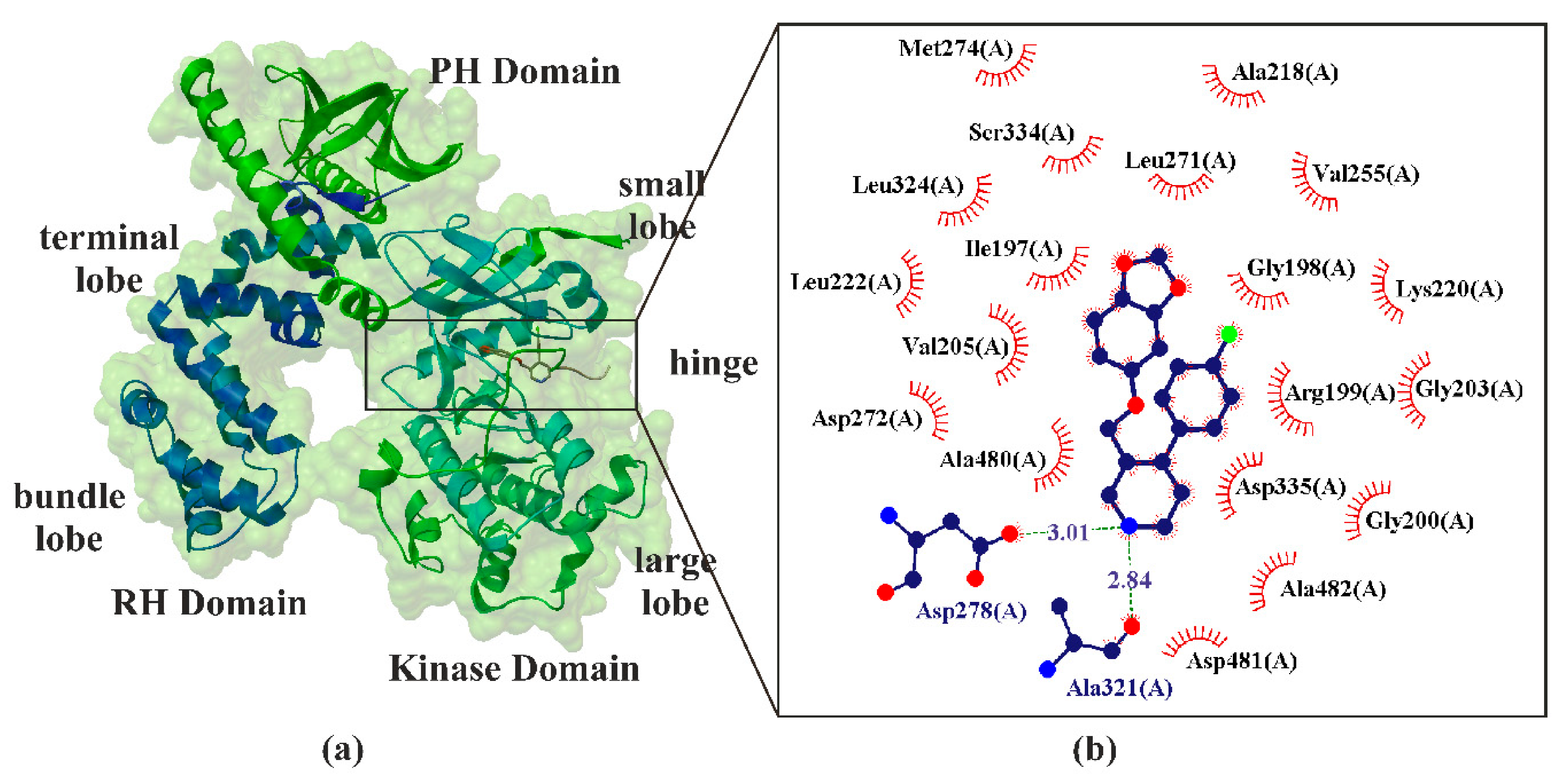
6. Paroxetine as Kinase GRK2 Inhibitors
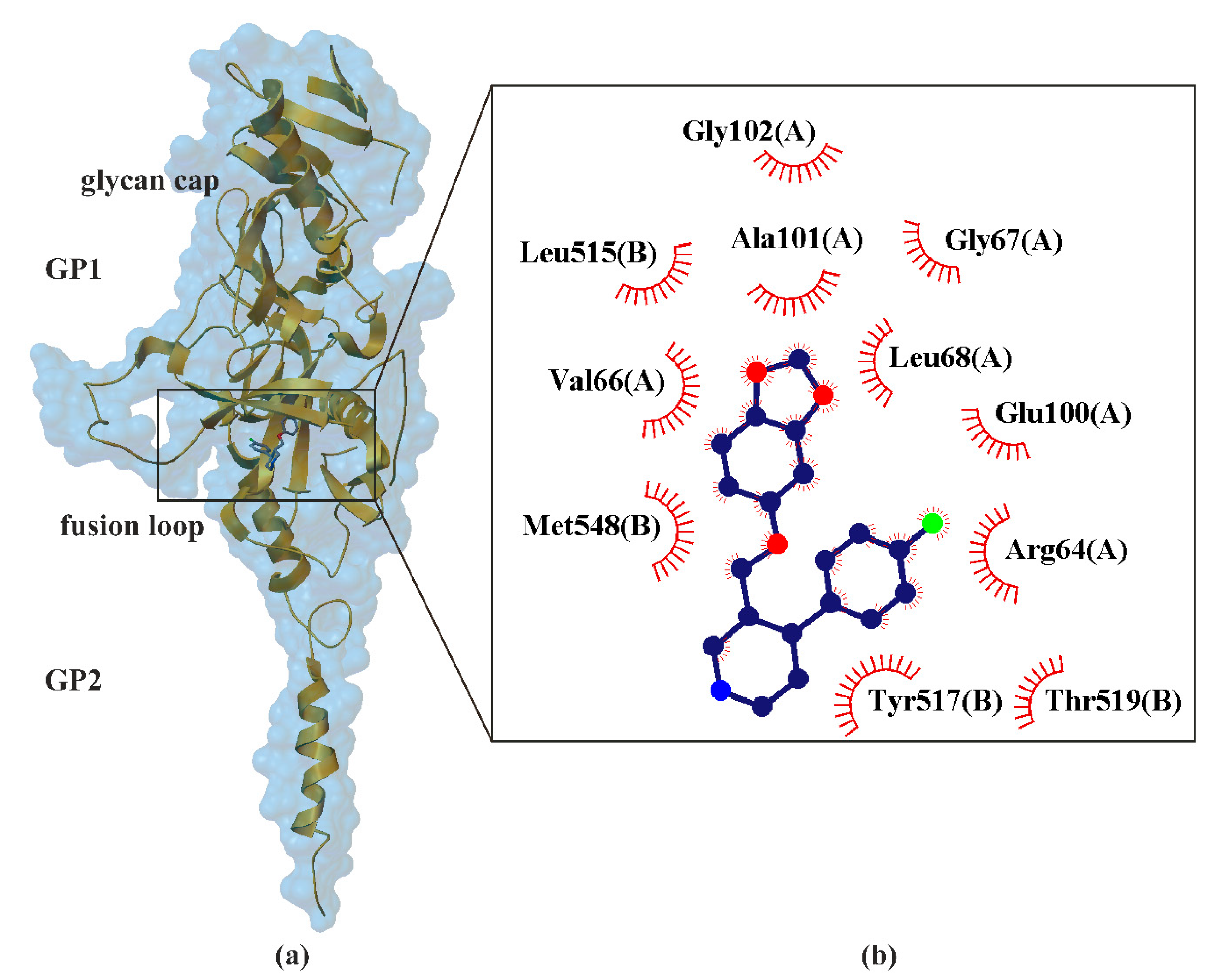
7. Paroxetine as Ebolavirus Inhibitors
8. Paroxetine as KIT and JAK Inhibitors
9. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Anxiety Disorders |
| ADAA | American Society of Anxiety and Depression |
| ABL | Abl Tyrosine Kinases |
| aLeuT | Aquifex Aeolicus Leucine Transporter |
| ATC code | Anatomical Therapeutic Chemical Classification System |
| ATP | Adenosine Triphosphate |
| BBB | Blood Brain Barrier |
| C-Src ABL | Src Tyrosine Kinases |
| CYP | Cytochromes P450 |
| CYP1A2 | Cytochrome P450 Family 1 Subfamily A Member 2 |
| CYP2B4 | Cytochrome P450 Family 2 Subfamily B Member 4 |
| CYP2B6 | Cytochrome P450 Family 2 Subfamily B Member 6 |
| CYP2C19 | Cytochrome P450 Family 2 Subfamily C Member 19 |
| CYP2C19 | Cytochrome P450 Family 2 Subfamily C Member 19 |
| CYP2D6 | Cytochrome P450 Family 2 Subfamily D Member 6 |
| CYP3A4 | Cytochrome P450 Family 3 Subfamily A Member 4 |
| CYP3A5 | Cytochrome P450 Family 3 Subfamily A Member 5 |
| DAT | Dopamine Transporter |
| dDAT | Drosophila Dopamine Transporter |
| EBOV GP | Ebolavirus Glycoprotein |
| EHF | Ebola Hemorrhagic Fever |
| EVD | Ebola Virus Disease |
| FDA | Food and Drug Administration |
| FYN | Fyn Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| GABA | γ-Aminobutyric Acid |
| GAD | Generalized Anxiety Disorder |
| GAT | γ-Aminobutyric Acid Transporter |
| GlyT | Glycine Transporter |
| GP | Glycoprotein |
| GPCR | G-Protein-Coupled Receptors |
| GRK | G-Protein-Coupled Receptors Kinase |
| GRK1 | G-Protein Coupled Receptor Kinase 1 |
| GRK2 | G-Protein Coupled Receptor Kinase 2 |
| GRK5 | G-Protein Coupled Receptor Kinase 5 |
| hDAT | Human Dopamine Transporter |
| HEM | Hemeproteins |
| hNET | Human Norepinephrine Transporter |
| hSERT | Human Serotonin Transporter |
| ICD-10-CM | International Classification of Diseases, Tenth Revision, Clinical Modification |
| JAK | Janus Kinase |
| Ki | Binding Affinity |
| KIT | Tyrosine-Kinase Inhibitor |
| L | Polymerase |
| LeuT | Leucine Transporter |
| MAT | Monoamine Transporters |
| MDD | Major Depressive Disorder |
| MDL | O-linked glycosylated mucin-like domain |
| MET | Met Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| NET | Norepinephrine Transporter |
| NP | Nucleoprotein |
| NSS | Neurotransmitter Sodium Symporter |
| OCD | Obsessive-Compulsive Disorder |
| PD | Panic Disorder |
| PDB ID | Protein Data Bank Identifier |
| PDD | Premenstrual Dysphoric Disorder |
| P-gl | P-Glycoprotein |
| PTSD | Posttraumatic Stress Disorder |
| RBD | Receptor Binding Domain |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| SAD | Social Anxiety Disorder |
| SERT | Serotonin Transporter |
| SLC6 | Solute Carrier 6 Family of Transporters |
| SRC | Src Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| SSRI | Selective Serotonin Reuptake Inhibitors |
| TM | Transmembrane Segment |
| VP24 | Viral Protein 24 |
| VP30 | Viral Protein 30 |
| VP35 | Viral Protein 35 |
| VP40 | Viral Protein 40 |
| βARK-1 | β-Adrenergic Receptor Kinase 1 |
References
- Gmitrowicz, A.; Janas-Kozik, M. Zaburzenia Psychiczne Dzieci i Młodzieży; Medical Tribune Polska: Warszawa, Poland, 2018; pp. 87–101. [Google Scholar]
- Unbound Medicine. F40–F48—Anxiety, Dissociative, Stress-Related, Somatoform and Other Nonpsychotic Mental Disorders. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/934326/all/F40_F48___Anxiety__dissociative__stress_related__somatoform_and_other_nonpsychotic_mental_disorders (accessed on 30 January 2021).
- Rockhill, C.; Kodish, I.; DiBattisto, C.; Macias, M.; Varley, C.; Ryan, S. Anxiety Disorders in Children and Adolescents. Curr. Probl. Pediatric Adolesc. Health Care 2010, 40, 66–99. [Google Scholar] [CrossRef]
- Adaa.org. Understand the Facts Anxiety and Depression Association of America. Available online: https://adaa.org/understanding-anxiety (accessed on 29 January 2021).
- Unbound Medicine.F40.11—Social Phobia, Generalized. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/943473/all/F40_11___Social_phobia_generalized?q=anxiety+disorder+social (accessed on 30 January 2021).
- West, K.B.; Wilbanks, J.; Suveg, C. Exposure Therapy for Separation Anxiety Disorder. Exposure Therapy for Children with Anxiety and OCD: Clinician’s Guide to Integrated Treatment; Academic Press: London, UK, 2020; pp. 143–153. [Google Scholar]
- Knappe, S.; Beesdo-Baum, K.; Fehm, L.; Lieb, R.; Wittchen, H. Characterizing the association between parenting and adolescent social phobia. J. Anxiety Disord. 2012, 26, 608–616. [Google Scholar] [CrossRef]
- Heimberg, R.; Hofmann, S.; Liebowitz, M.; Schneier, F.; Smits, J.; Stein, M.; Hinton, D.; Craske, M. Social anxiety disorder in DSM-5. Depression and Anxiety. Depress. Anxiety 2014, 31, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Bazzano, A.T.; Mangione-Smith, R.; Schonlau, M.; Suttorp, M.J.; Brook, R.H. Off-label prescribing to children in the United States outpatient setting. Acad. Pediatrics 2009, 9, 81–88. [Google Scholar] [CrossRef] [PubMed]
- ATC Code. Anatomical Therapeutic Chemical Classification System. Available online: http://www.atccode.com/ (accessed on 30 January 2021).
- DrugBank. Paroxetine. Available online: https://go.drugbank.com/drugs/DB00715 (accessed on 30 January 2021).
- Sharma, S.; Dang, S. Paroxetine loaded PLGA nanoparticles. Mater. Today Proc. 2020, 28, 205–210. [Google Scholar] [CrossRef]
- Tang, S.W.; Helmeste, D. Paroxetine. Expert Opin. Pharmacother. 2008, 9, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.J.; Viera, A.J.; Parman, C.L. Off-label applications for SSRIs. Am. Fam. Physician 2003, 68, 498–504. [Google Scholar] [PubMed]
- Skånland, S.S.; Cieślar-Pobuda, A. Off-label uses of drugs for depression. Eur. J. Pharmacol. 2019, 865, 172732. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–Protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Unbound Medicine. F41.1—Generalized Anxiety Disorder. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/905183/all/F41_1___Generalized_anxiety_disorder?q=300.02 (accessed on 30 January 2021).
- Unbound Medicine. 300.3—Obsessive-Compulsive Disorders. Available online: https://www.unboundmedicine.com/icd//search?st=OSS&q=300.3 (accessed on 30 January 2021).
- Unbound Medicine. F33—Major Depressive Disorder, Recurrent. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/930155/all/F33___Major_depressive_disorder_recurrent?q=depressive+disorder+major (accessed on 30 January 2021).
- Unbound Medicine. F32.81—Premenstrual Dysphoric Disorder. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/968287/all/F32_81___Premenstrual_dysphoric_disorder?q=disorder+dysphoric+premenstrual (accessed on 30 January 2021).
- Unbound Medicine. F43.1—Post-Traumatic Stress Disorder (PTSD). Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/929135/all/F43_1___Post_traumatic_stress_disorder__PTSD_?q=f43.1 (accessed on 30 January 2021).
- Unbound Medicine. F41.0—Panic Disorder [Episodic Paroxysmal Anxiety]. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/915404/all/F41_0___Panic_disorder_[episodic_paroxysmal_anxiety]?q=anxiety+disorder+generalized (accessed on 30 January 2021).
- Liu, X.; Li, X.; Zhang, C.; Sun, M.; Sun, Z.; Xu, Y.; Tian, X. Efficacy and tolerability of fluvoxamine in adults with social anxiety disorder: A meta-analysis. Medicine 2018, 97, e11547. [Google Scholar] [CrossRef]
- Bandelow, B.; Sher, L.; Bunevicius, R.; Hollander, E.; Kasper, S.; Zohar, J.; Möller, H. Guidelines for the pharmacological treatment of anxiety disorders, obsessive–compulsive disorder and posttraumatic stress disorder in primary care. Int. J. Psychiatry Clin. Pract. 2012, 16, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.S.; Anderson, I.M.; Nutt, D.J.; Bandelow, B.; Bond, A.; Davidson, J.R.; den Boer, J.A.; Fineberg, N.A.; Knapp, M.S.J.; Hu, W. Evidence-based guidelines for the pharmacological treatment of anxiety disorders: Recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2005, 19, 567–596. [Google Scholar] [CrossRef] [PubMed]
- Yonkers, K.A.; Gullion, C.; Williams, A.; Novak, K.; Rush, A.J. Paroxetine as a treatment for premenstrual dysphoric disorder. J. Clin. Psychopharmacol. 1996, 16, 3–8. [Google Scholar] [CrossRef]
- Aghakhani, K.S.S.; Farhidnia, N.; Fallah, F. Successful Suicide in a Child: Depression-Related or ParoxetineInduced? Int. J. Med Toxicol. Forensic Med. 2016, 6, 242–246. [Google Scholar]
- Lenox, R.H.; Frazer, A. Neuropsychopharmacology: The Fifth Generation of Progress; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2010; pp. 1139–1163. [Google Scholar]
- Schneeweiss, S.; Patrick, A.R.; Solomon, D.H.; Dormuth, C.R.; Miller, M.; Mehta, J.; Lee, J.C.; Wang, P.S. Comparative safety of antidepressant agents for children and adolescents regarding suicidal acts. Pediatrics 2010, 125, 876–888. [Google Scholar] [CrossRef]
- Teicher, M.H.; Glod, C.; Cole, J.O. Emergence of intense suicidal preoccupation during fluoxetine treatment. Am. J. Psychiatry 1990, 147, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, S.E.; McKenzie, J.E.; Cox, G.R.; Simmons, M.B.; Merry, S.N. Newer generation antidepressants for depressive disorders in children and adolescents. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, M.; Radice, S.; Clementi, E.; Molteni, M.; Nobile, M. Antidepressants and, suicide and self-injury: Causal or casual association? Int. J. Psychiatry Clin. Pract. 2016, 20, 47–51. [Google Scholar] [CrossRef]
- Morrison, J.; Schwartz, T.L. Adolescent angst or true intent? Suicidal behavior, risk, and neurobiological mechanisms in depressed children and teenagers taking antidepressants. Int. J. Emerg. Ment. Health 2014, 16, 247–250. [Google Scholar] [PubMed]
- Giner, L.; Nichols, C.M.; Zalsman, G.; Oquendo, M.A. Selective serotonin reuptake inhibitors and the risk for suicidality in adolescents: An update. Int. J. Adolesc. Med. Health 2005, 17, 211–220. [Google Scholar] [CrossRef]
- McKeown, R.E.; Cuffe, S.P.; Schulz, R.M. US suicide rates by age group, 1970–2002: An examination of recent trends. Am. J. Public Health 2006, 96, 1744–1751. [Google Scholar] [CrossRef]
- Hjalmarsson, L.; Corcos, M.; Jeammet, P. Selective serotonin reuptake inhibitors in major depressive disorder in children and adolescents (ratio of benefits/risks). Encephale 2005, 31, 309–316. [Google Scholar] [CrossRef]
- Hammad, T.A.; Laughren, T.; Racoosin, J. Suicidality in pediatric patients treated with antidepressant drugs. Arch. Gen. Psychiatry 2006, 63, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Termorshuizen, F.; Palmen, S.J.; Heerdink, E.R. Suicide behavior before and after the start with antidepressants: A high persistent risk in the first month of treatment among the young. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. Antidepressants may double risk of suicide and aggression in children, study finds. BMJ 2016, 352, i545. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.H. The treatment of child and adolescent depression: A matter of concern? Acta Psychiatr. Scand. 2007, 115, 169–170. [Google Scholar] [CrossRef]
- Li, X.; Hou, Y.; Su, Y.; Liu, H.; Zhang, B.; Fang, S. Efficacy and tolerability of paroxetine in adults with social anxiety disorder: A meta-analysis of randomized controlled trials. Medicine 2020, 99, e19573. [Google Scholar] [CrossRef]
- Nevels, R.M.; Gontkovsky, S.T.; Williams, B.E. Paroxetine—the antidepressant from hell? Probably not, but caution required. Psychopharmacol. Bull. 2016, 46, 77. [Google Scholar]
- Germann, D.; Ma, G.; Han, F.; Tikhomirova, A. Paroxetine Hydrochloride. Profiles of Drug Substances. Excip. Relat. Methodol. 2013, 38, 367–406. [Google Scholar]
- Agrawal, N.; Marco-Peiró, S.; Esteve-Romero, J.; Durgbanshi, A.; Bose, D.; Peris-Vicente, J.; Carda-Broch, S. Determination of paroxetine in blood and urine using micellar liquid chromatography with electrochemical detection. J. Chromatogr. Sci. 2014, 52, 1217–1223. [Google Scholar] [CrossRef]
- Pae, C.-U.; Patkar, A.A. Paroxetine: Current status in psychiatry. Expert Rev. Neurother. 2007, 7, 107–120. [Google Scholar] [CrossRef]
- Green, B. Focus on paroxetine. Curr. Med Res. Opin. 2003, 19, 13–21. [Google Scholar] [CrossRef]
- GlaxoSmithKline. PAXIL (Paroxetine Hydrochloride) Tablets and Oral Suspension: Prescribing Information. Available online: https://www.baumhedlundlaw.com/documents/pdf/dolin-trial-exhibits/JX-3-2007-Paxil-label-prescribing-information.pdf (accessed on 30 January 2021).
- GSK Canada. PAXIL—Product monograph. Compend. Pharm. Specialties. Ott. Can. Pharm. Assoc. 2014, 1, 1255–1260. [Google Scholar]
- Patetsos, E.; Horjales-Araujo, E. Treating Chronic Pain with SSRIs: What Do We Know? Pain Res. Manag. 2016. [Google Scholar] [CrossRef]
- Uttamsingh, V.; Gallegos, R.; Liu, J.F.; Harbeson, S.L.; Bridson, G.W.; Cheng, C.; Wells, D.S.; Graham, P.B.; Zelle, R.; Tung, R. Altering Metabolic Profiles of Drugs by Precision Deuteration: Reducing Mechanism-Based Inhibition of CYP2D6 by Paroxetines. J. Pharmacol. Exp. Ther. 2015, 354, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; de Souza Dantas, L.M.; Crowell-Davis, S.L. Selective serotonin reuptake inhibitors. Vet. Psychopharmacol. 2019, 103–128. [Google Scholar]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Gouaux, E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. [Google Scholar] [CrossRef]
- Coleman, J.A.; Navratna, V.; Antermite, D.; Yang, D.; Bull, J.A.; Gouaux, E. Chemical and structural investigation of the paroxetine-human serotonin transporter complex. eLife 2020, 9, e56427. [Google Scholar] [CrossRef]
- Shah, M.B.; Kufareva, I.; Pascual, J.; Zhang, Q.; Stout, C.D.; Halpert, J.R. A structural snapshot of CYP2B4 in complex with paroxetine provides insights into ligand binding and clusters of conformational states. J. Pharmacol. Exp. Ther. 2013, 346, 113–120. [Google Scholar] [CrossRef]
- Homan, K.T.; Wu, E.; Wilson, M.W.; Singh, P.; Larsen, S.D.; Tesmer, J.J. Structural and functional analysis of G protein–coupled receptor kinase inhibition by paroxetine and a rationally designed analog. Mol. Pharmacol. 2014, 85, 237–248. [Google Scholar] [CrossRef]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Goehring, A.; Wang, K.H.; Penmatsa, A.; Ressler, R.; Gouaux, E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 2013, 503, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhao, Y.; Fry, E.E.; Stuart, D.I. Target identification and mode of action of four chemically divergent drugs against Ebolavirus infection. J. Med. Chem. 2018, 61, 724–733. [Google Scholar] [CrossRef] [PubMed]
- GlobalData UK Ltd. Post-Traumatic Stress Disorder (PTSD)—Opportunity Analysis and Forecasts to 2028. Available online: https://store.globaldata.com/report/gdhc095poa--post-traumatic-stress-disorder-ptsd-opportunity-analysis-and-forecasts-to-2028/?utm_source=pharma&utm_medium=Research%20Reports&utm_campaign=RSReportPage?utm_source=pharma&utm_campaign=RShomepage (accessed on 30 January 2021).
- PubChem. Paroxetine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/43815 (accessed on 30 January 2021).
- CCDC. Access Structures. Available online: https://www.ccdc.cam.ac.uk/structures/ (accessed on 30 January 2021).
- Pina, M.F.; Pinto, J.F.; Sousa, J.J.; Fábián, L.; Zhao, M.; Craig, D.Q. Identification and characterization of stoichiometric and nonstoichiometric hydrate forms of paroxetine HCl: Reversible changes in crystal dimensions as a function of water absorption. Mol. Pharm. 2012, 9, 3515–3525. [Google Scholar] [CrossRef]
- Yokota, M.; Uekusa, H.; Ohashi, Y. Structure analyses of two crystal forms of paroxetine hydrochloride. Bull. Chem. Soc. Jpn. 1999, 72, 1731–1736. [Google Scholar] [CrossRef]
- SpectraBase. Paroxetine. Available online: https://spectrabase.com/spectrum/3GQhXHaQFxC (accessed on 30 January 2021).
- Salsbury, J.S.; Isbester, P.K. Quantitative 1H NMR method for the routine spectroscopic determination of enantiomeric purity of active pharmaceutical ingredients fenfluramine, sertraline, and paroxetine. Magn. Reson. Chem. 2005, 43, 910–917. [Google Scholar] [CrossRef]
- MassBank of North America (MoNA). Spectrum AU152606 for Paroxetine. Available online: https://mona.fiehnlab.ucdavis.edu/spectra/display/AU152606 (accessed on 30 January 2021).
- Schrapp, A.; Mory, C.; Duflot, T.; Pereira, T.; Imbert, L.; Lamoureux, F. The right blood collection tube for therapeutic drug monitoring and toxicology screening procedures: Standard tubes, gel or mechanical separator? Clin. Chim. Acta 2018, 488, 196–201. [Google Scholar] [CrossRef]
- Kaye, C.M.; Haddock, R.E.; Langley, P.F.; Mellows, G.; Tasker, T.C.G.; Zussman, B.D.; Greb, W.H. A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr. Scand. 1989, 80, 60–75. [Google Scholar] [CrossRef]
- Davis, B.; Nagarajan, A.; Forrest, L.; Singh, S. Mechanism of Paroxetine (Paxil) Inhibition of the Serotonin Transporter. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- HMDB. Showing Metabocard for Paroxetine (HMDB0014853). Available online: https://hmdb.ca/metabolites/HMDB0014853#identification (accessed on 30 January 2021).
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- KEGG. DRUG: Paroxetine. Available online: https://www.kegg.jp/dbget-bin/www_bget?dr:D02362 (accessed on 30 January 2021).
- Human Cytochrome P450s. Available online: https://drnelson.uthsc.edu/human.P450.table.html (accessed on 30 January 2021).
- Channels, D.B. Available online: https://webchemdev.ncbr.muni.cz/ChannelsDB/ (accessed on 30 January 2021).
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Mäeenpäeä, J.; Taavitsainen, P.; Rautio, A.; Raunio, H. Inhibition and induction of human cytochrome P450 (CYP) enzymes. Xenobiotica 1998, 28, 1203–1253. [Google Scholar] [CrossRef]
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Haddock, R.; Johnson, A.; Langley, P.; Nelson, D.; Pope, J.; Thomas, D.; Woods, F. Metabolic pathway of paroxetine in animals and man and the comparative pharmacological properties of its metabolites. Acta Psychiatr. Scand. 1989, 80, 24–26. [Google Scholar] [CrossRef]
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: A review. Cns Drug Rev. 2001, 7, 25–47. [Google Scholar] [CrossRef]
- Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes 2020, 11, 1295. [Google Scholar] [CrossRef]
- Gay, S.C.; Roberts, A.G.; Halpert, J.R. Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR. Future Med. Chem. 2010, 2, 1451–1468. [Google Scholar] [CrossRef]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand access channels in cytochrome P450 enzymes: A review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef]
- Poulos, T.L. Cytochrome P450: Molecular architecture, mechanism, and prospects for rational inhibitor design. Pharm. Res. 1988, 5, 67–75. [Google Scholar] [CrossRef]
- Yosca, T.H.; Ledray, A.P.; Ngo, J.; Green, M.T. A new look at the role of thiolate ligation in cytochrome P450. Jbic J. Biol. Inorg. Chem. 2017, 22, 209–220. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R.; De Voss, J.J. Oxidizing species in the mechanism of cytochrome P450. Nat. Prod. Rep. 2002, 19, 477–493. [Google Scholar] [CrossRef]
- Kamel, E.M.; Lamsabhi, A.M. The quasi-irreversible inactivation of cytochrome P450 enzymes by paroxetine: A computational approach. Org. Biomol. Chem. 2020, 18, 3334–3345. [Google Scholar] [CrossRef]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metab. Pers. Ther. 2012, 27, 185–197. [Google Scholar] [CrossRef]
- Wang, H.; Tompkins, L.M. CYP2B6: New insights into a historically overlooked cytochrome P450 isozyme. Curr. Drug Metab. 2008, 9, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.A.; de Montellano, P.R.O. Inhibition of cytochrome P450 enzymes. In Cytochrome P450; Springer: Berlin/Heidelberg, Germany, 2005; pp. 247–322. [Google Scholar]
- Stahl, S.M. Essential Psychopharmacology: Neuroscientific Basis and Practical Applications; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Walsky, R.L.; Astuccio, A.V.; Obach, R.S. Evaluation of 227 Drugs for In Vitro Inhibition of Cytochrome P450 2B6. Drug Metab. 2006, 7, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-W.; Kim, E.-J.; Nyiramana, M.M.; Shin, E.-J.; Jin, H.; Ryu, J.H.; Kang, K.R.; Lee, G.-W.; Kim, H.J.; Han, J. Paroxetine induces apoptosis of human breast cancer mcf-7 cells through Ca2+-and p38 map kinase-dependent ros generation. Cancers 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hsieh, J.-H.; Huang, R.; Sakamuru, S.; Hsin, L.-Y.; Xia, M.; Shockley, K.R.; Auerbach, S.; Kanaya, N.; Lu, H. Cell-based high-throughput screening for aromatase inhibitors in the Tox21 10K library. Toxicol. Sci. 2015, 147, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, D.; Hsin, L.-Y.; Kanaya, N.; Wong, C.; Yip, R.; Sakamuru, S.; Xia, M.; Yuan, Y.-C.; Witt, K. AroER tri-screen is a biologically relevant assay for endocrine disrupting chemicals modulating the activity of aromatase and/or the estrogen receptor. Toxicol. Sci. 2014, 139, 198–209. [Google Scholar] [CrossRef]
- Rowland, P.; Blaney, F.E.; Smyth, M.G.; Jones, J.J.; Leydon, V.R.; Oxbrow, A.K.; Lewis, C.J.; Tennant, M.G.; Modi, S.; Eggleston, D.S. Crystal structure of human cytochrome P450 2D6. J. Biol. Chem. 2006, 281, 7614–7622. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Raunio, H.; Pelkonen, O. The functional role of CYP2B6 in human drug metabolism: Substrates and inhibitors in vitro, in vivo and in silico. Curr. Drug Metab. 2006, 7, 705–714. [Google Scholar] [CrossRef]
- Zhou, S.-F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Wilderman, R.P.; Halpert, R.J. Plasticity of CYP2B enzymes: Structural and solution biophysical methods. Curr. Drug Metab. 2012, 13, 167–176. [Google Scholar] [CrossRef]
- Ravna, A.W.; Sylte, I.; Dahl, S.G. Structure and localisation of drug binding sites on neurotransmitter transporters. J. Mol. Modeling 2009, 15, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Navratna, V.; Gouaux, E. Insights into the mechanism and pharmacology of neurotransmitter sodium symporters. Curr. Opin. Struct. Biol. 2019, 54, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef]
- Andersen, J.; Ringsted, K.B.; Bang-Andersen, B.; Strømgaard, K.; Kristensen, A.S. Binding site residues control inhibitor selectivity in the human norepinephrine transporter but not in the human dopamine transporter. Sci. Rep. 2015, 5, 15650. [Google Scholar] [CrossRef]
- Cool, D.R.; Leibach, F.H.; Ganapathy, V. High-affinity paroxetine binding to the human placental serotonin transporter. Am. J. Physiol. Cell Physiol. 1990, 259, C196–C204. [Google Scholar] [CrossRef]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef]
- Hoehn-Saric, R.; Merchant, A.F.; Keyser, M.L. Effects of Clonidine on Anxiety Disorders. Arch. Gen. Psychiatry 1981, 38, 1278–1282. [Google Scholar] [CrossRef]
- Leerssen, E.C.M.; Tak, R.O.; Breur, J.M. Severe transient neonatal long QT syndrome due to maternal paroxetine usage: A case report. Cardiol. Young 2019, 29, 1300–1301. [Google Scholar] [CrossRef]
- Patel, N.; Wiśniowska, B.; Jamei, M.; Polak, S. Real Patient and its Virtual Twin: Application of Quantitative Systems Toxicology Modelling in the Cardiac Safety Assessment of Citalopram. Aaps J. 2018, 20, 6. [Google Scholar] [CrossRef]
- Rannversson, H.; Andersen, J.; Bang-Andersen, B.; Strømgaard, K. Mapping the binding site for escitalopram and paroxetine in the human serotonin transporter using genetically encoded photo-cross-linkers. ACS Chem. Biol. 2017, 12, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, L.; Andersen, J.; Thomsen, M.; Hansen, S.M.; Zhao, X.; Sandelin, A.; Strømgaard, K.; Kristensen, A.S. Interaction of Antidepressants with the Serotonin and Norepinephrine Transporters mutational studies of the S1 substrate binding pocket. J. Biol. Chem. 2012, 287, 43694–43707. [Google Scholar] [CrossRef] [PubMed]
- Abramyan, A.M.; Slack, R.D.; Meena, S.; Davis, B.A.; Newman, A.H.; Singh, S.K.; Shi, L. Computation-guided analysis of paroxetine binding to hSERT reveals functionally important structural elements and dynamics. Neuropharmacology 2019, 161, 107411. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Irwin, J.J. Docking screens: Right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Dorn, G.W. GRK mythology: G-protein receptor kinases in cardiovascular disease. J. Mol. Med. 2009, 87, 455–463. [Google Scholar] [CrossRef]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Port, J.D.; Bristow, M.R. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J. Mol. Cell Cardiol. 2001, 33, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204. [Google Scholar] [CrossRef]
- Sutherland, E.W.; Robison, G.A.; Butcher, R.W. Some aspects of the biological role of adenosine 3’,5’-monophosphate (cyclic AMP). Circulation 1968, 37, 279–306. [Google Scholar] [CrossRef]
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Hullmann, J.; Traynham, C.J.; Coleman, R.C.; Koch, W.J. The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res. 2016, 110, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef]
- Waldschmidt, H.V.; Homan, K.T.; Cato, M.C.; Cruz-Rodríguez, O.; Cannavo, A.; Wilson, M.W.; Song, J.; Cheung, J.Y.; Koch, W.J.; Tesmer, J.J. Structure-based design of highly selective and potent G protein-coupled receptor kinase 2 inhibitors based on paroxetine. J. Med. Chem. 2017, 60, 3052–3069. [Google Scholar] [CrossRef]
- Raake, P.W.; Vinge, L.E.; Gao, E.; Boucher, M.; Rengo, G.; Chen, X.; DeGeorge, B.R.J.; Matkovich, S.; Houser, S.R.; Most, P. G protein–coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ. Res. 2008, 103, 413–422. [Google Scholar] [CrossRef]
- Lodowski, D.T.; Barnhill, J.F.; Pyskadlo, R.M.; Ghirlando, R.; Sterne-Marr, R.; Tesmer, J.J. The role of Gβγ and domain interfaces in the activation of G protein-coupled receptor kinase 2. Biochemistry 2005, 44, 6958–6970. [Google Scholar] [CrossRef] [PubMed]
- Murga, C.; Arcones, A.C.; Cruces-Sande, M.; Briones, A.M.; Salaices, M.; Mayor, F.J. G protein-coupled receptor kinase 2 (GRK2) as a potential therapeutic target in cardiovascular and metabolic diseases. Front. Pharmacol. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Unbound Medicine. A98.4—Ebola Virus Disease. Available online: https://www.unboundmedicine.com/icd/view/ICD-10-CM/888247/all/Ebola%20(virus) (accessed on 30 January 2021).
- World Health Organization. Ebola Virus Disease. Available online: https://www.who.int/en/news-room/fact-sheets/detail/ebola-virus-disease (accessed on 30 January 2021).
- Na, W.; Park, N.; Yeom, M.; Song, D. Ebola outbreak in Western Africa 2014: What is going on with Ebola virus? Clin. Exp. Vaccine Res. 2015, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Jagga, S.; Sharma, A.R.; Chakraborty, C.; Lee, S.-S. Ebola virus disease: Recent advances in diagnostics and therapeutics. Asian Pac. J. Trop. Med. 2019, 12, 385. [Google Scholar]
- Hartman, A.L.; Bird, B.H.; Towner, J.S.; Antoniadou, Z.-A.; Zaki, S.R.; Nichol, S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 2008, 82, 2699–2704. [Google Scholar] [CrossRef]
- Yu, D.-S.; Weng, T.-H.; Wu, X.-X.; Wang, F.X.; Lu, X.-Y.; Wu, H.-B.; Wu, N.-P.; Li, L.-J.; Yao, H.-P. The lifecycle of the Ebola virus in host cells. Oncotarget 2017, 8, 55750. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef]
- Nehls, J.; Businger, R.; Hoffmann, M.; Brinkmann, C.; Fehrenbacher, B.; Schaller, M.; Maurer, B.; Schönfeld, C.; Kramer, D.; Hailfinger, S. Release of immunomodulatory Ebola virus glycoprotein-containing microvesicles is suppressed by tetherin in a species-specific manner. Cell Rep. 2019, 26, 1841–1853.e6. [Google Scholar] [CrossRef]
- Rooney, A.; Grant, R. Pharmacological treatment of depression in patients with a primary brain tumour. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Jang, W.J.; Jung, S.K.; Vo, T.T.L.; Jeong, C.H. Anticancer activity of paroxetine in human colon cancer cells: Involvement of MET and ERBB3. J. Cell Mol. Med. 2019, 23, 1106–1115. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Fiordelisi, A.; Gambardella, J.; Franco, A.; del Giudice, C.; Oliveti, M. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020, 7, 1571–1584. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, H.; Wang, H. Inverse in silico–in vitro fishing of unexpected paroxetine kinase targets from tumor druggable kinome. J. Mol. Modeling 2020, 26, 1–9. [Google Scholar] [CrossRef]
- Hao, Z.; Sadek, I. Sunitinib: The antiangiogenic effects and beyond. Oncotargets Ther. 2016, 9, 5495. [Google Scholar] [CrossRef]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007, 6, 734–745. [Google Scholar] [CrossRef]
- Plante, J.; Eason, C.; Snyder, A.; Elston, D. Tofacitinib in the treatment of lichen planopilaris: A retrospective review. J. Am. Acad. Dermatol. 2020, 83, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- FDA. Drug Approval Package. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-936_Paxil.cfm (accessed on 30 January 2021).
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiao, F.; Feng, Z.; Li, M.; Kong, L.; Huang, L.; Li, H.; Liu, F.; Zhang, H.; Zhang, W. Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Kalra, S.; Kaushal, S. Systematic review of tofacitinib: A new drug for the management of rheumatoid arthritis. Clin. Ther. 2014, 36, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tofacitinib: A review in rheumatoid arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, X.; Feng, M.; Wang, Q.; Zhou, P. Gaussian process: A promising approach for the modeling and prediction of peptide binding affinity to MHC proteins. Protein Pept. Lett. 2011, 18, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. Aaps J. 2012, 14, 759–763. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | Target | Resolution | Organism | Reference |
|---|---|---|---|---|
| 5I6X, 5I6Z | SERT | 3.14 Å, 4.53 Å | Homo sapiens, Mus musculus | [52] |
| 6AWN | SERT | 3.62 Å | Homo sapiens, Mus musculus | [53] |
| 6VRH | SERT | 3.30 Å | Homo sapiens, Mus musculus | [54] |
| 4JLT | P450 2B4 | 2.14 Å | Oryctolagus cuniculus | [55] |
| 4L9I | GRK1 | 2.32 Å | Bos taurus | [56] |
| 3V5W | GRK2 | 2.07 Å | Bos taurus, Homo sapiens | [57] |
| 4MM4 | LeuBAT | 2.89 Å | Aquifex aeolicus VF5 | [58] |
| 6F6I | EBOV GP | 2.40 Å | Ebola virus | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalska, M.; Nowaczyk, J.; Fijałkowski, Ł.; Nowaczyk, A. Paroxetine—Overview of the Molecular Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 1662. https://doi.org/10.3390/ijms22041662
Kowalska M, Nowaczyk J, Fijałkowski Ł, Nowaczyk A. Paroxetine—Overview of the Molecular Mechanisms of Action. International Journal of Molecular Sciences. 2021; 22(4):1662. https://doi.org/10.3390/ijms22041662
Chicago/Turabian StyleKowalska, Magdalena, Jacek Nowaczyk, Łukasz Fijałkowski, and Alicja Nowaczyk. 2021. "Paroxetine—Overview of the Molecular Mechanisms of Action" International Journal of Molecular Sciences 22, no. 4: 1662. https://doi.org/10.3390/ijms22041662
APA StyleKowalska, M., Nowaczyk, J., Fijałkowski, Ł., & Nowaczyk, A. (2021). Paroxetine—Overview of the Molecular Mechanisms of Action. International Journal of Molecular Sciences, 22(4), 1662. https://doi.org/10.3390/ijms22041662