
Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
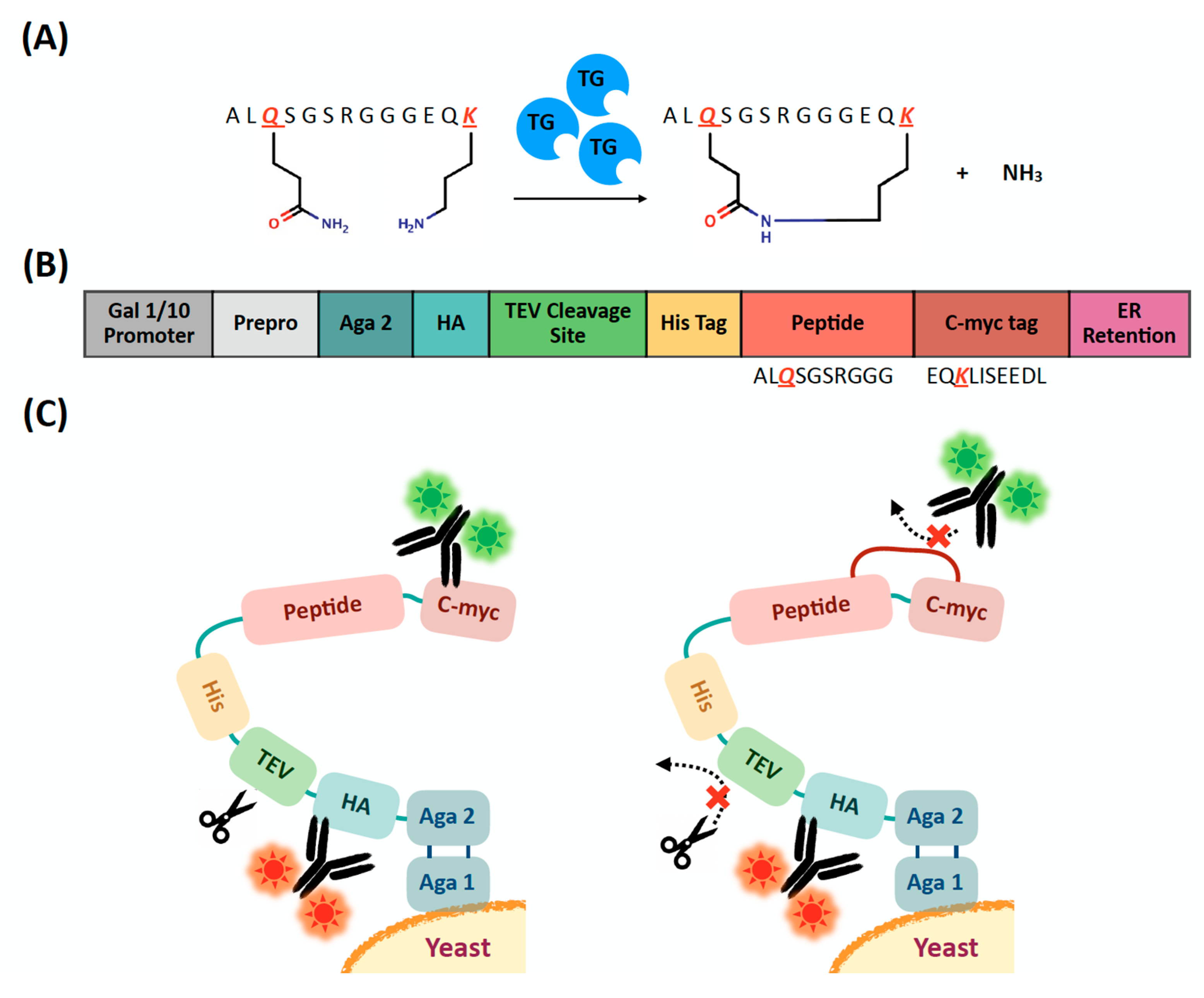
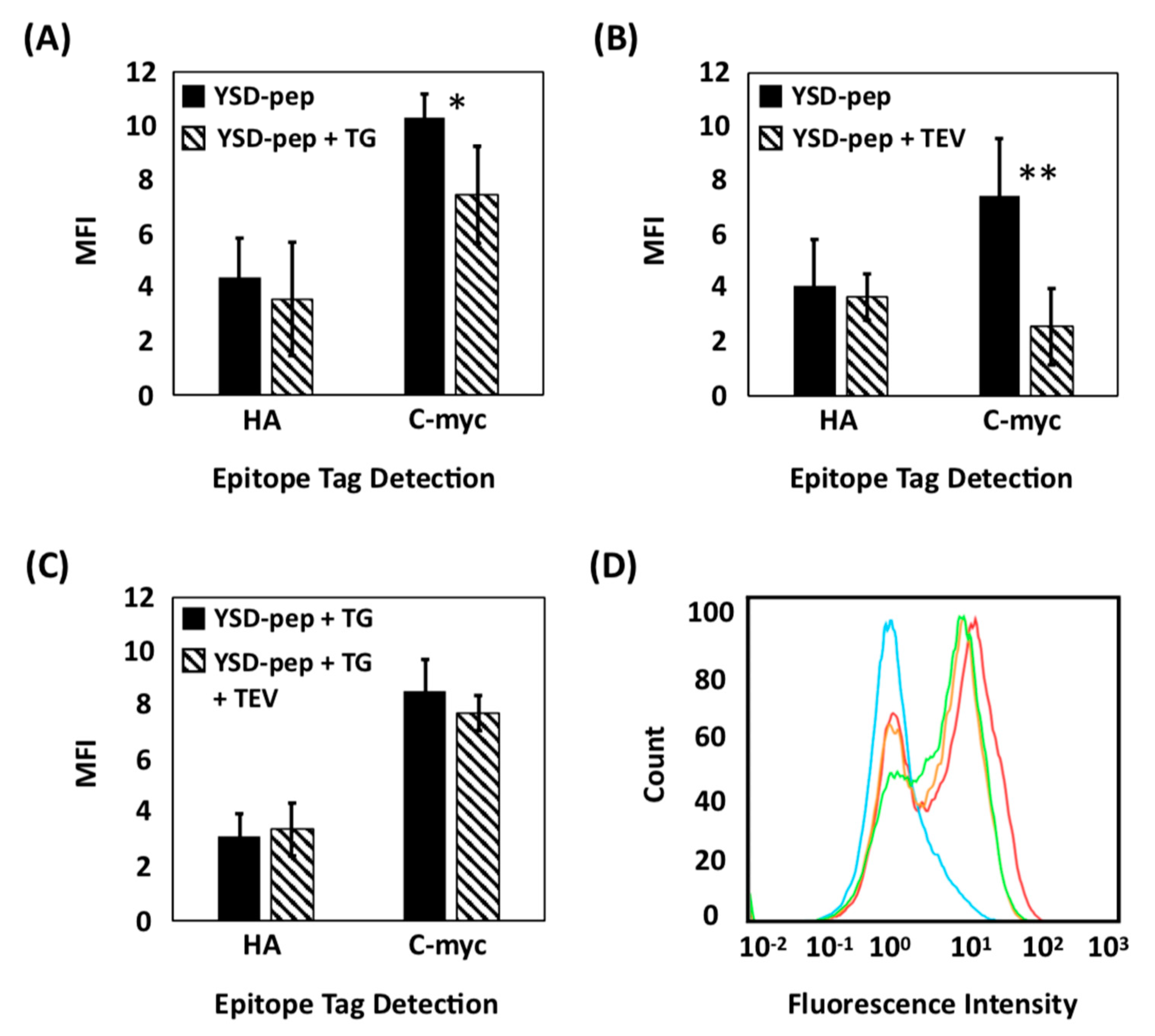
2.1. Evaluation of Extracellular Transglutaminase-Mediated Putative Cyclization of Yeast-Displayed Peptides
2.2. Design, Construction, and Screening of a Yeast Display Library of Peptides Modified Using Extracellular Transglutaminase
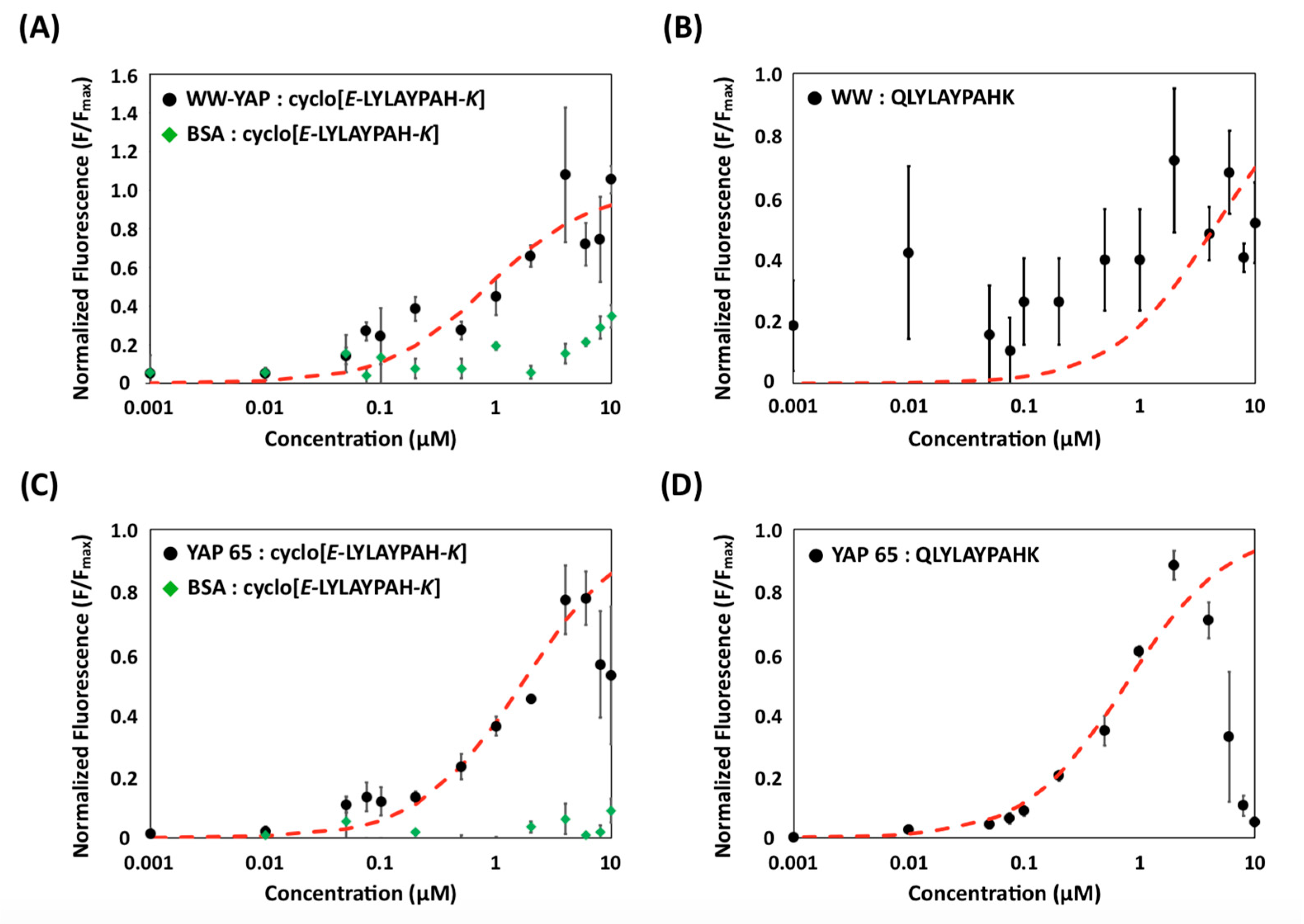
2.3. Evaluation of Binding Affinity and Specificity of (Putative) Cyclo[E-LYLAYPAH-K] via Yeast Surface Titrations
2.4. Evaluation of Binding Affinity and Specificity of Cyclo[E-LYLAYPAH-K] via Static Binding Assays on Chromatographic Resins
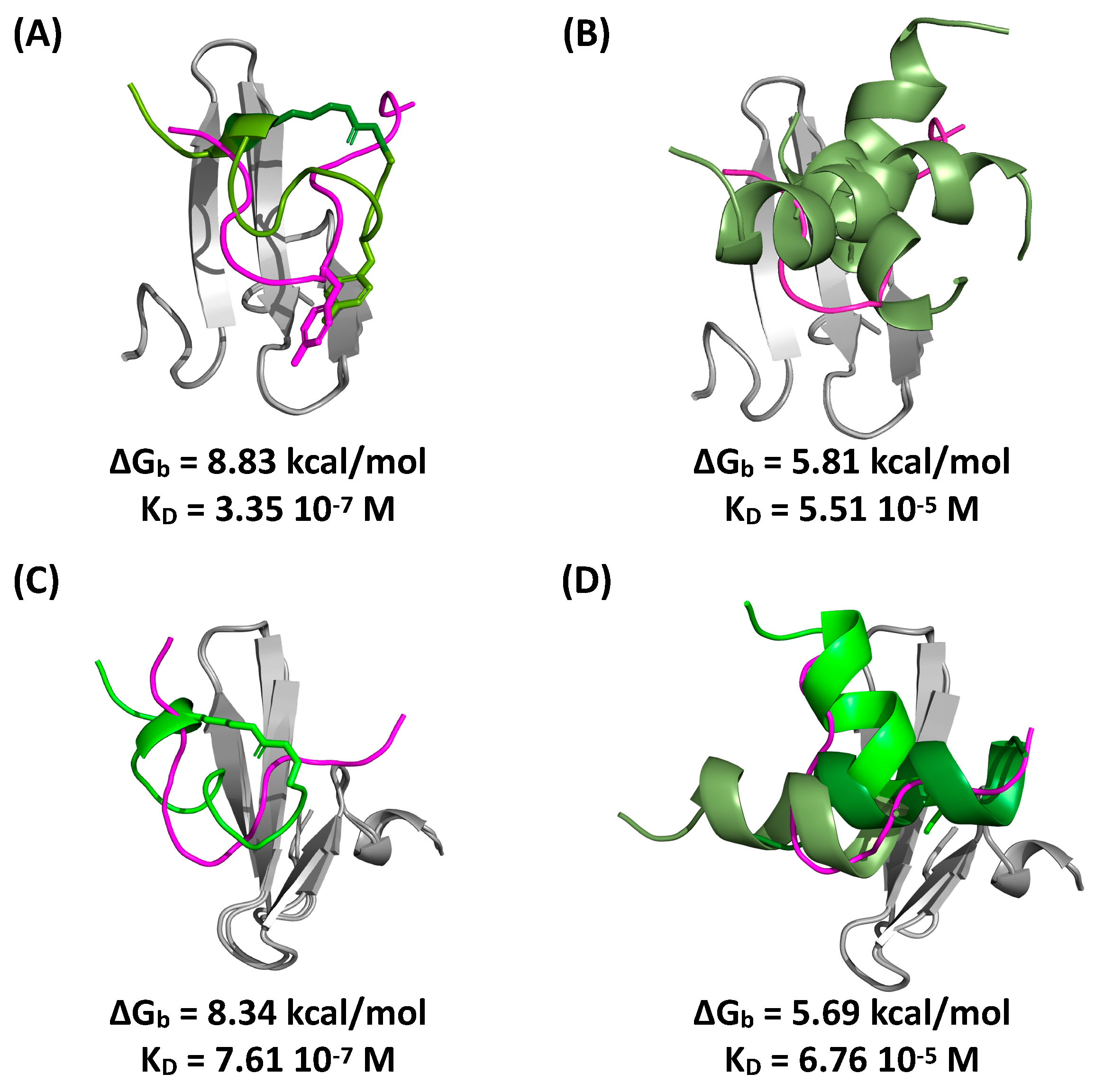
2.5. Evaluation of Peptide Binding via Molecular Docking and Dynamics Simulations
2.6. Evaluation of Peptide Cyclization in Yeast Cells by Intracellular Transglutaminase
3. Materials and Methods
3.1. Plasmids and Yeast Cell Culture
3.2. Plasmid Construction for Yeast Surface Display of Linear Peptides
3.3. Extracellular Treatment of Yeast Cells Displaying Linear Peptide Precursors with Transglutaminase
3.4. Evaluation of Peptide Cyclization Mediated by Extracellular Transglutaminase via Flow Cytometry
3.5. Expression, Purification, and Biotinylation of Recombinant N-Terminal YAP, WW Domains of YAP, and TEV
3.6. Construction of Yeast Display Library of Linear Peptide Precursors for Putative Cyclization by Extracellular Transglutaminase
3.7. Screening of a Yeast Display Library of Linear Peptide Precursors Putatively Cyclized via Extracellular Transglutaminase to Identify Cyclic Peptide Binders for N-Terminal YAP and Its WW Domains
3.8. Estimation of Binding Affinity via Yeast Surface Titrations
3.9. Synthesis of Peptides Cyclo[E-LYLAYPAH-K] and RYSPPPPYSSHS (PTCH) on Toyopearl Resin
3.10. Evaluation of Binding Capacity, Affinity, and Selectivity of Peptide Functionalized Resin for N-Terminal YAP and WW-YAP
3.11. In Silico Modeling of WW:Peptide Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bird, G.H.; Madani, N.; Perry, A.F.; Princiotto, A.M.; Supko, J.G.; He, X.; Gavathiotis, E.; Sodroski, J.G.; Walensky, L.D. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. USA 2010, 107, 14093–14098. [Google Scholar] [CrossRef]
- Szewczuk, Z.; Gibbs, B.F.; Yue, S.Y.; Purisima, E.O.; Konishi, Y. Conformationally restricted thrombin inhibitors resistant to proteolytic digestion. Biochemistry 1992, 31, 9132–9140. [Google Scholar] [CrossRef]
- Horswill, A.R.; Benkovic, S.J. Cyclic peptides, a chemical genetics tool for biologists. Cell Cycle 2005, 4, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the Reality. Chem. Biol. 2014, 18, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, P.; Berlicki, Ł. Peptide-based inhibitors of protein-protein interactions. Bioorg. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef]
- Roberts, R.W.; Szostak, J. RNA-peptide fusions for the in vitro selection of peptides. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Lam, K.I.T.S.; Liu, R.; Miyamoto, S.; Lehman, A.L.; Tuscano, J.M. Applications of One-Bead One-Compound Libraries and Chemical Microarrays in Signal Transduction Research. Acc. Chem. Res. 2003, 36, 370–377. [Google Scholar] [CrossRef]
- Krumpe, L.R.H.; Mori, T. Potential of phage-displayed peptide library technology to identify functional targeting peptides. Expert Opin. Drug Discov. 2007, 2, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Keefe, A.D.; Szostak, J.W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 3750–3755. [Google Scholar] [CrossRef]
- Linciano, S.; Pluda, S.; Bacchin, A.; Angelini, A. Molecular evolution of peptides by yeast surface display technology. Medchemcomm 2019, 10, 1569–1580. [Google Scholar] [CrossRef]
- Pfaff, M.; Tangemann, K.; Müller, B.; Gurrath, M.; Müller, G.; Kessler, H.; Timpl, R.; Engel, J. Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J. Biol. Chem. 1994, 269, 20233–20238. [Google Scholar] [CrossRef]
- Roxin, Á.; Zheng, G. Flexible or fixed: A comparative review of linear and cyclic cancer-targeting peptides. Future Med. Chem. 2012, 4, 1601–1618. [Google Scholar] [CrossRef]
- McLafferty, M.A.; Kent, R.B.; Ladner, R.C.; Markland, W. M13 bacteriophage displaying disulfide-constrained microproteins. Gene 1993, 128, 29–36. [Google Scholar] [CrossRef]
- O’Neil, K.T.; Hoess, R.H.; Jackson, S.A.; Ramachandran, N.S.; Mousa, S.A.; DeGrado, W.F. Identification of novel peptide antagonists for GPIIb/IIIa from a conformationally constrained phage peptide library. Proteins Struct. Funct. Genet. 1992, 14, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lupold, S.E.; Rodriguez, R. Disulfide-constrained peptides that bind to the extracellular portion of the prostate-specific membrane antigen. Mol. Cancer Ther. 2004, 3, 597–603. [Google Scholar]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfidebond stability. Methods Enzymol. 1995, 251, 8–28. [Google Scholar] [PubMed]
- Millward, S.W.; Takahashi, T.T.; Roberts, R.W. A General Route for Post-Translational Cyclization of mRNA Display Libraries. J. Am. Chem. Soc. 2005, 127, 14142–14143. [Google Scholar] [CrossRef] [PubMed]
- Millward, S.W.; Fiacco, S.; Austin, R.J.; Roberts, R.W. Design of cyclic peptides that bind protein surfaces with antibody-like affinity. ACS Chem. Biol. 2007, 2, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Bacon, K.; Blain, A.; Burroughs, M.; McArthrur, N.; Rao, B.M.; Menegatti, S. Isolation of Chemically Cyclized Peptide Binders Using Yeast Surface Display. ACS Comb. Sci. 2020, 22, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Wetzel, R. Novel cyclization chemistry especially suited for biologically derive unprotected peptides. Int. J. Pept. Protein Res. 1992, 39, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Van Der Donk, W.A. Ribosomally synthesized and post-translationally modified peptide natural products: New insights into the role of leader and core peptides during biosynthesis. Chem. A Eur. J. 2013, 19, 7662–7677. [Google Scholar] [CrossRef]
- Touati, J.; Angelini, A.; Hinner, M.J.; Heinis, C. Enzymatic cyclisation of peptides with a transglutaminase. ChemBioChem 2011, 12, 38–42. [Google Scholar] [CrossRef]
- Repka, L.M.; Chekan, J.R.; Nair, S.K.; Van Der Donk, W.A. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem. Rev. 2017, 117, 5457–5520. [Google Scholar] [CrossRef]
- Tsukiji, S.; Nagamune, T. Sortase-mediated ligation: A gift from gram-positive bacteria to protein engineering. ChemBioChem 2009, 10, 787–798. [Google Scholar] [CrossRef]
- Antos, J.M.; Popp, M.W.; Ernst, R.; Chew, G.; Spooner, E.; Ploegh, H.L. A Straight Path to Circular Proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.K.T.; Kam, A.; Loo, S.; Jansson, A.E.; Pan, L.X.; Tam, J.P. Butelase 1: A Versatile Ligase for Peptide and Protein Macrocyclization. J. Am. Chem. Soc. 2015, 137, 15398–15401. [Google Scholar] [CrossRef]
- Flühe, L.; Knappe, T.A.; Gattner, M.J.; Schäfer, A.; Burghaus, O.; Linne, U. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 2012, 8, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; Burnier, J.P.; Wells, J.A. Enzymatic Cyclization of Linear Peptide Esters Using Subtiligase. J. Am. Chem. Soc. 1995, 117, 819–820. [Google Scholar] [CrossRef]
- Bordusa, F. Enzymes for Peptide Cyclization. ChemBioChem 2001, 2, 405–409. [Google Scholar] [CrossRef]
- Xu, M.Q.; Evans, T.C. Intein-mediated ligation and cyclization of expressed proteins. Methods 2001, 24, 257–277. [Google Scholar] [CrossRef]
- Osher, E.L.; Tavassoli, A. Intracellular Production of Cyclic Peptide Libraries with SICLOPPS. Methods Mol. Biol. 2017, 1495, 27–39. [Google Scholar] [CrossRef]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef]
- Tavassoli, A.; Benkovic, S.J. Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli. Nat. Protoc. 2007, 2, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, K.J.; Walker, M.C.; Van Der Donk, W.A. Development and Application of Yeast and Phage Display of Diverse Lanthipeptides. ACS Cent. Sci. 2018, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.H.; Moosmeier, M.A.; Aumüller, T.; Thein, M.; Bosma, T.; Rink, R.; Groth, K.; Zulley, M.; Siegers, K.; Tissot, K.; et al. Phage display and selection of lanthipeptides on the carboxy-terminus of the gene-3 minor coat protein. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Bosma, T.; Kuipers, A.; Bulten, E.; de Vries, L.; Rink, R.; Moll, G.N. Bacterial display and screening of posttranslationally thioether-stabilized peptides. Appl. Environ. Microbiol. 2011, 77, 6794–6801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Javitt, G.; Ben-Barak-Zelas, Z.; Jerabek-Willemsen, M.; Fishman, A. Constitutive expression of active microbial transglutaminase in Escherichia coli and comparative characterization to a known variant. BMC Biotechnol. 2017, 17, 23. [Google Scholar] [CrossRef]
- Yi, L.; Gebhard, M.C.; Li, Q.; Taft, J.M.; Georgiou, G.; Iverson, B.L. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc. Natl. Acad. Sci. USA 2013, 110, 7229–7234. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Rachel, N.; Pelletier, J. Biotechnological Applications of Transglutaminases. Biomolecules 2013, 3, 870–888. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.J.L.M.; Van Maarseveen, J.H.; Nuijens, T. Enzyme-catalyzed peptide cyclization. Drug Discov. Today Technol. 2017, 26, 11–16. [Google Scholar] [CrossRef]
- Deweid, L.; Neureiter, L.; Englert, S.; Schneider, H.; Deweid, J.; Yanakieva, D.; Sturm, J.; Bitsch, S.; Christmann, A.; Avrutina, O.; et al. Directed Evolution of a Bond-Forming Enzyme: Ultrahigh-Throughput Screening of Microbial Transglutaminase Using Yeast Surface Display. Chem. A Eur. J. 2018, 24, 15195–15200. [Google Scholar] [CrossRef]
- Sugimura, Y.; Hosono, M.; Wada, F.; Yoshimura, T.; Maki, M.; Hitomi, K. Screening for the Preferred Substrate Sequence of Transglutaminase Using a Phage-displayed Peptide Library. J. Biol. Chem. 2006, 281, 17699–17706. [Google Scholar] [CrossRef]
- Iglesias-Bexiga, M.; Castillo, F.; Cobos, E.S.; Oka, T.; Sudol, M.; Luque, I. WW domains of the yes-kinase-associated-protein (YAP) transcriptional regulator behave as independent units with different binding preferences for PPxY motif-containing ligands. PLoS ONE 2015, 10, e0113828. [Google Scholar] [CrossRef] [PubMed]
- Aragón, E.; Goerner, N.; Xi, Q.; Gomes, T.; Gao, S.; Massagué, J.; MacIas, M.J. Structural basis for the versatile interactions of Smad7 with regulator WW domains in TGF-β pathways. Structure 2012, 20, 1726–1736. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Columbus, J.; Zhang, Y.; Wu, D.; Lian, L.; Yang, S.; Goodwin, J.; Luczak, C.; Carter, M.; Chen, L.; et al. A map of WW domain family interactions. Proteomics 2004, 4, 643–655. [Google Scholar] [CrossRef]
- Gera, N.; Hussain, M.; Rao, B.M. Protein selection using yeast surface display. Methods 2013, 60, 15–26. [Google Scholar] [CrossRef]
- Chao, G.; Lau, W.L.; Hackel, B.J.; Sazinsky, S.L.; Lippow, S.M.; Wittrup, K.D. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 2006, 1, 755–768. [Google Scholar] [CrossRef]
- Bacon, K.; Burroughs, M.; Blain, A.; Menegatti, S.; Rao, B.M. Screening Yeast Display Libraries against Magnetized Yeast Cell Targets Enables Efficient Isolation of Membrane Protein Binders. ACS Comb. Sci. 2019, 21, 817–832. [Google Scholar] [CrossRef]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. Des. Sel. 2001, 14, 993–1000. [Google Scholar] [CrossRef]
- Benatuil, L.; Perez, J.M.; Belk, J.; Hsieh, C.-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010, 23, 155–159. [Google Scholar] [CrossRef]
- Gera, N.; Hussain, M.; Wright, R.C.; Rao, B.M. Highly stable binding proteins derived from the hyperthermophilic Sso7d scaffold. J. Mol. Biol. 2011, 409, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [PubMed]
- Chandra, K.; Roy, T.K.; Shalev, D.E.; Loyter, A.; Gilon, C.; Gerber, R.B.; Friedler, A. A Tandem In Situ Peptide Cyclization through Trifluoroacetic Acid Cleavage. Angew. Chem. Int. Ed. 2014, 53, 9450–9455. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color Test for the Detection of Free Terminal Amino Groups in the Solid-Phase Synthesis of Peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Menegatti, S.; Ward, K.L.; Naik, A.D.; Kish, W.S.; Blackburn, R.K.; Carbonell, R.G. Reversible Cyclic Peptide Libraries for the Discovery of Affinity Ligands. Anal. Chem. 2013, 85, 9229–9237. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tomillero, I.; Rodriguez, H.; Albericio, F. Tetrahydropyranyl, a Nonaromatic Acid-Labile Cys Protecting Group for Fmoc Peptide Chemistry. Org. Lett. 2015, 17, 1680–1683. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-1: Protein Preparation Wizard; Epik, Schrödinger, LLC.; Impact, Schrödinger, LLC.; Prime, Schrödinger, LLC.: New York, NY, USA, 2020.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Version 1.2.0. Available online: http://avogadro.cc/ (accessed on 15 December 2020).
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the opls force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Fu, J.; Yang, H.; Wang, J. Computational design of the helical hairpin structure of membrane-active antibacterial peptides based on RSV glycoprotein epitope scaffold. Comput. Biol. Chem. 2018, 73, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Yu, H.; Lin, Y.S. Toward structure prediction of cyclic peptides. Phys. Chem. Chem. Phys. 2015, 17, 4210–4219. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Cheatham, T.E.I.; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Quimbar, P.; Malik, U.; Sommerhoff, C.P.; Kaas, Q.; Chan, L.Y.; Huang, Y.H.; Grundhuber, M.; Dunse, K.; Craik, D.J.; Anderson, M.A.; et al. High-affinity cyclic peptide matriptase inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Spiliotopoulos, D.; Kastritis, P.L.; Melquiond, A.S.J.; Bonvin, A.M.J.J.; Musco, G.; Rocchia, W.; Spitaleri, A. dMM-PBSA: A New HADDOCK Scoring Function for Protein-Peptide Docking. Front. Mol. Biosci. 2016, 3, 46. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Ligand | KD (μM) | ||
|---|---|---|---|
| WW-YAP | N-YAP | BSA | |
| cyclo[E-LYLAYPAH-K] | 0.68 ± 0.21 | 1.75 ± 0.33 | N/D |
| QLYLAYPAHK | L/B | 0.74 ± 0.15 | N/A |
| Peptide Ligand | Target Protein | |||
|---|---|---|---|---|
| WW-Yap | N-YAP | |||
| KD (μM) | Qmax (mg/mL) | KD (μM) | Qmax (mg/mL) | |
| cyclo[E-LYLAYPAH-K] | 2.64 ± 0.71 | 5.60 ± 0.42 | 0.34 ± 0.02 | 3.03 ± 0.13 |
| PTCH peptide | 2.88 ± 0.48 | 8.3 ± 0.27 | 0.20 ± 0.07 | 4.11 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowen, J.; Schneible, J.; Bacon, K.; Labar, C.; Menegatti, S.; Rao, B.M. Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands. Int. J. Mol. Sci. 2021, 22, 1634. https://doi.org/10.3390/ijms22041634
Bowen J, Schneible J, Bacon K, Labar C, Menegatti S, Rao BM. Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands. International Journal of Molecular Sciences. 2021; 22(4):1634. https://doi.org/10.3390/ijms22041634
Chicago/Turabian StyleBowen, John, John Schneible, Kaitlyn Bacon, Collin Labar, Stefano Menegatti, and Balaji M. Rao. 2021. "Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands" International Journal of Molecular Sciences 22, no. 4: 1634. https://doi.org/10.3390/ijms22041634
APA StyleBowen, J., Schneible, J., Bacon, K., Labar, C., Menegatti, S., & Rao, B. M. (2021). Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands. International Journal of Molecular Sciences, 22(4), 1634. https://doi.org/10.3390/ijms22041634