
Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties


Abstract
1. Introduction
2. Properties of Conformationally Constrained Peptides
3. Constrainment Strategies and Chemistries
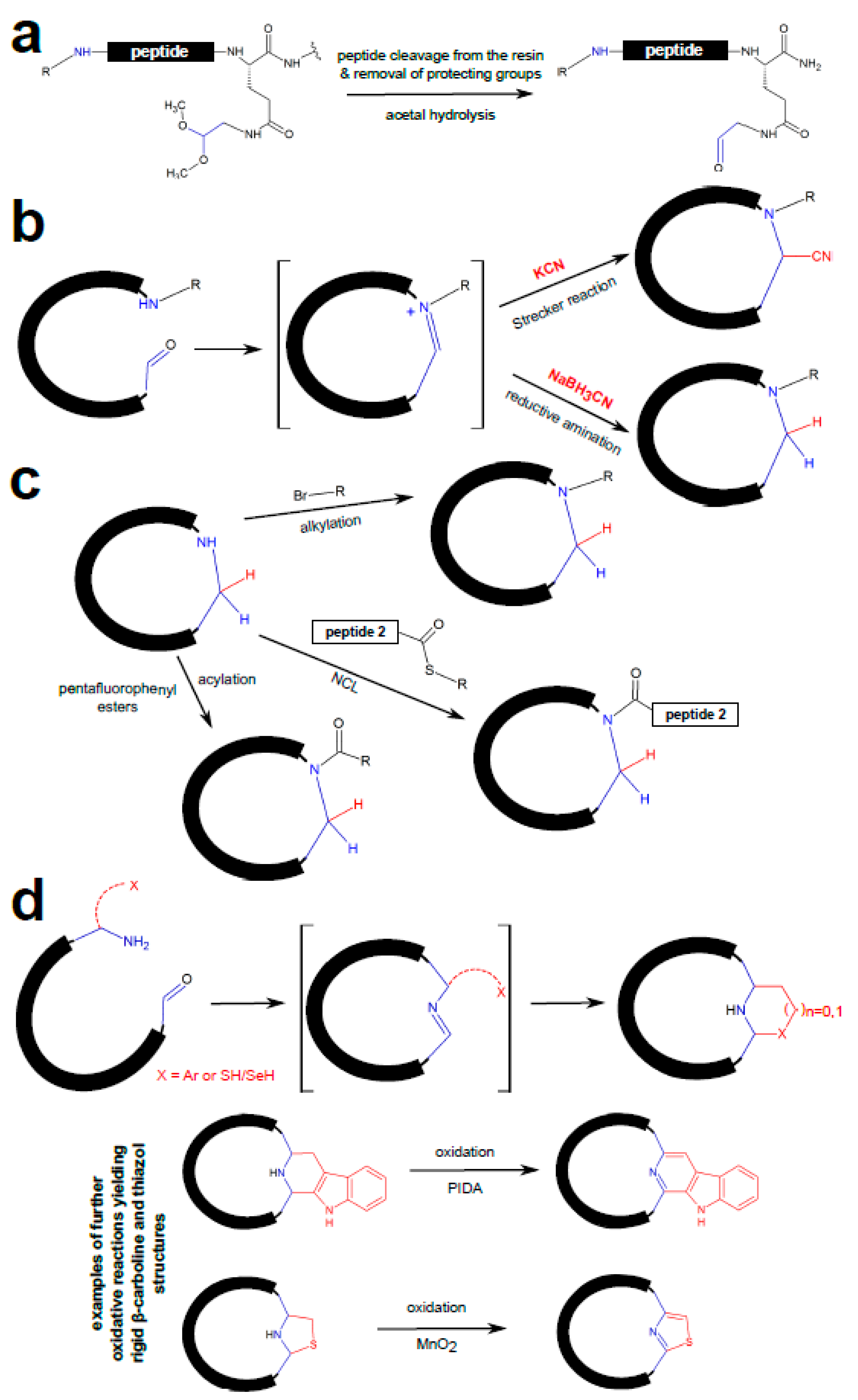
3.1. Chemical Peptide Ligation and Bridging
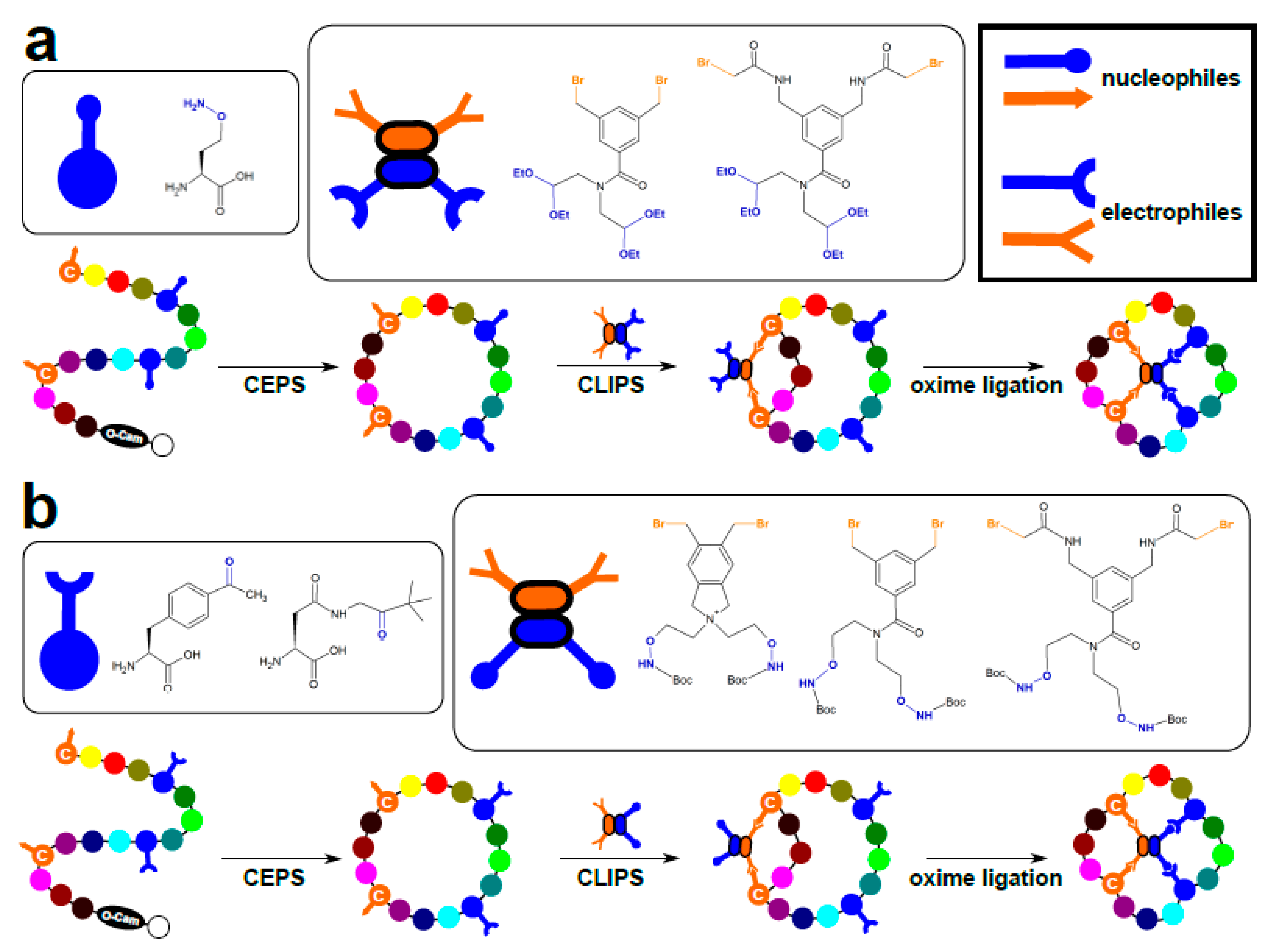
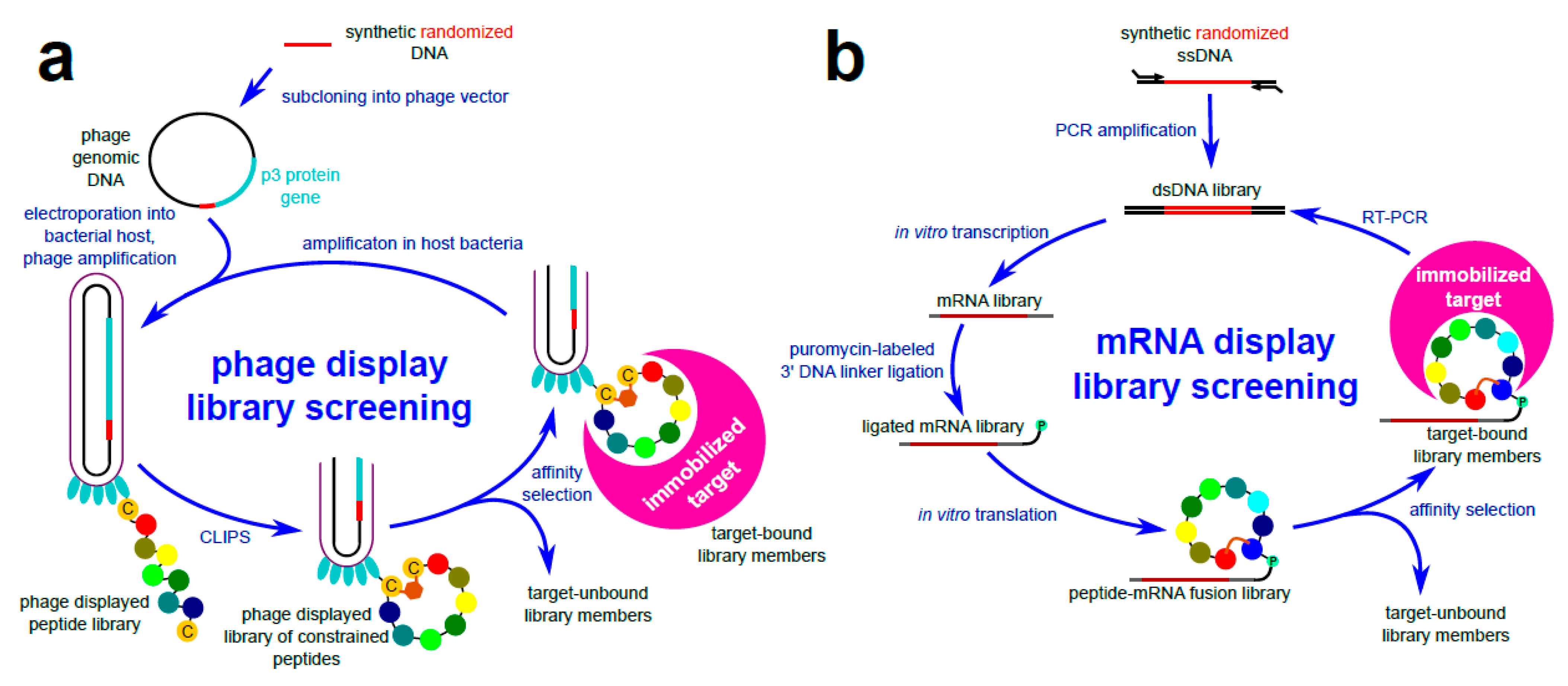
3.2. Chemical Ligation of Peptides onto Scaffolds (CLIPS)
3.3. Enzymatic Peptide Cyclization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | N-Terminal Recognition Sequence | C-Terminal Recognition Sequence (Cleavage Site Denoted with Caron Sign) | Ring Sizes (Residues) | Advantages | Disadvantages |
|---|---|---|---|---|---|
| sortase A | (G)n | LPXTˇG | ≥16 | commercially available, recombinant, high ligation yield | LPXT(G)n footprint, reaction reversible, competing oligomerization, moderate catalytic efficiency (0.1–1 molar eq. required) |
| butelase 1 | X1X2, where X1 is not P, D, or E, and X2 is L, I, V or C | (N/D)ˇHV | ≥10 | broad substrate specificity (thus minimal footprint), high ligation yield, high catalytic efficiency (~0.005 molar eq. required), reaction irreversible | only accessible through isolation, very few residues tolerated in position X2 |
| peptiligases [omniligase] | any dipeptide lacking P | essentially any tetrapeptide without C-terminal P; requires activated C-terminal carboxy group (ester) | ≥13 | very broad substrate specificity (hence no footprint), extraordinary catalytic efficiency (only ~0.0003 molar eq. required), reaction irreversible, high yield, [tolerates co-solvents and chaotropic agents, commercially available, compatible with non-peptidic backbone moieties] | requires activated C-terminal carboxy group (ester) |
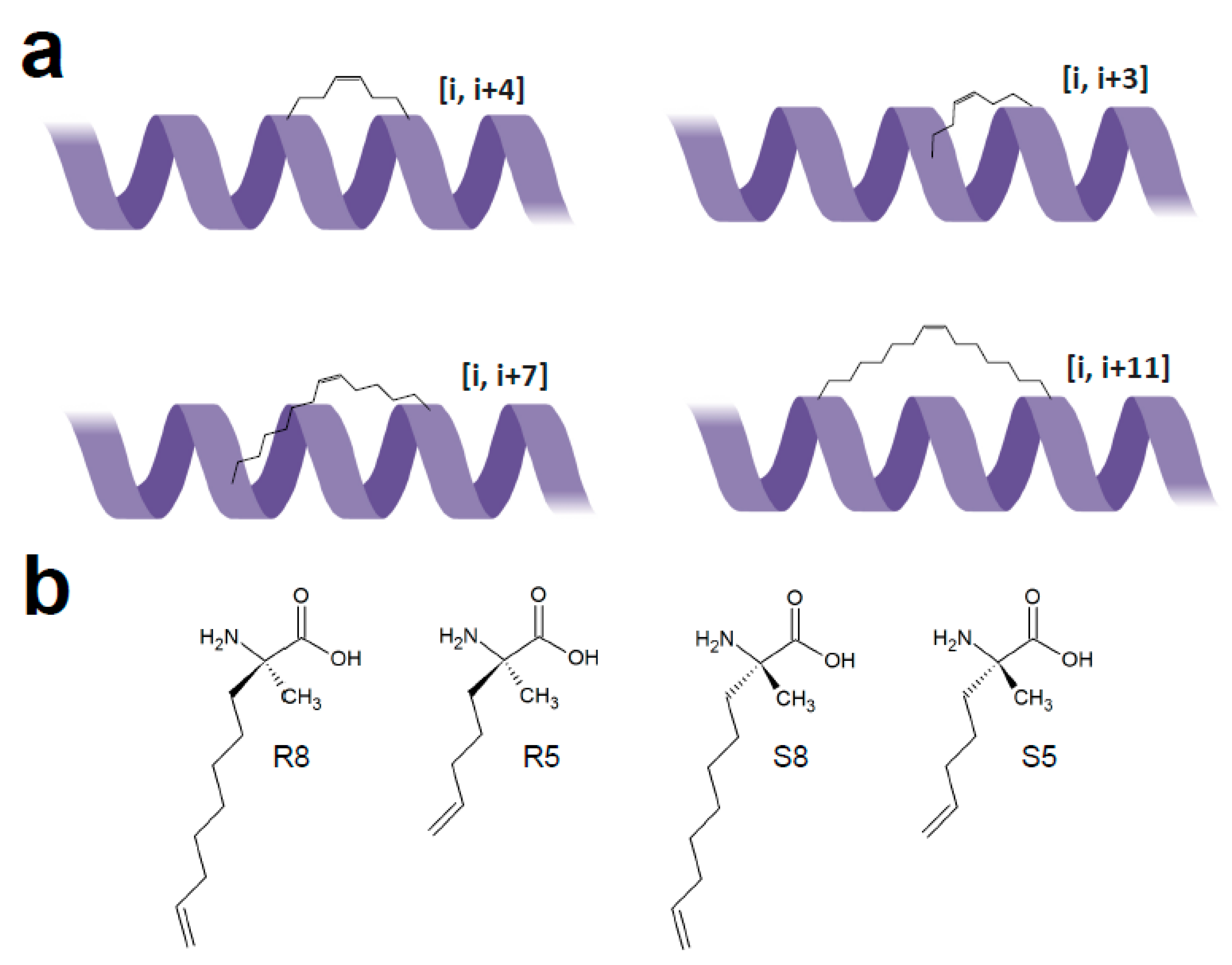
3.4. Peptide Stapling
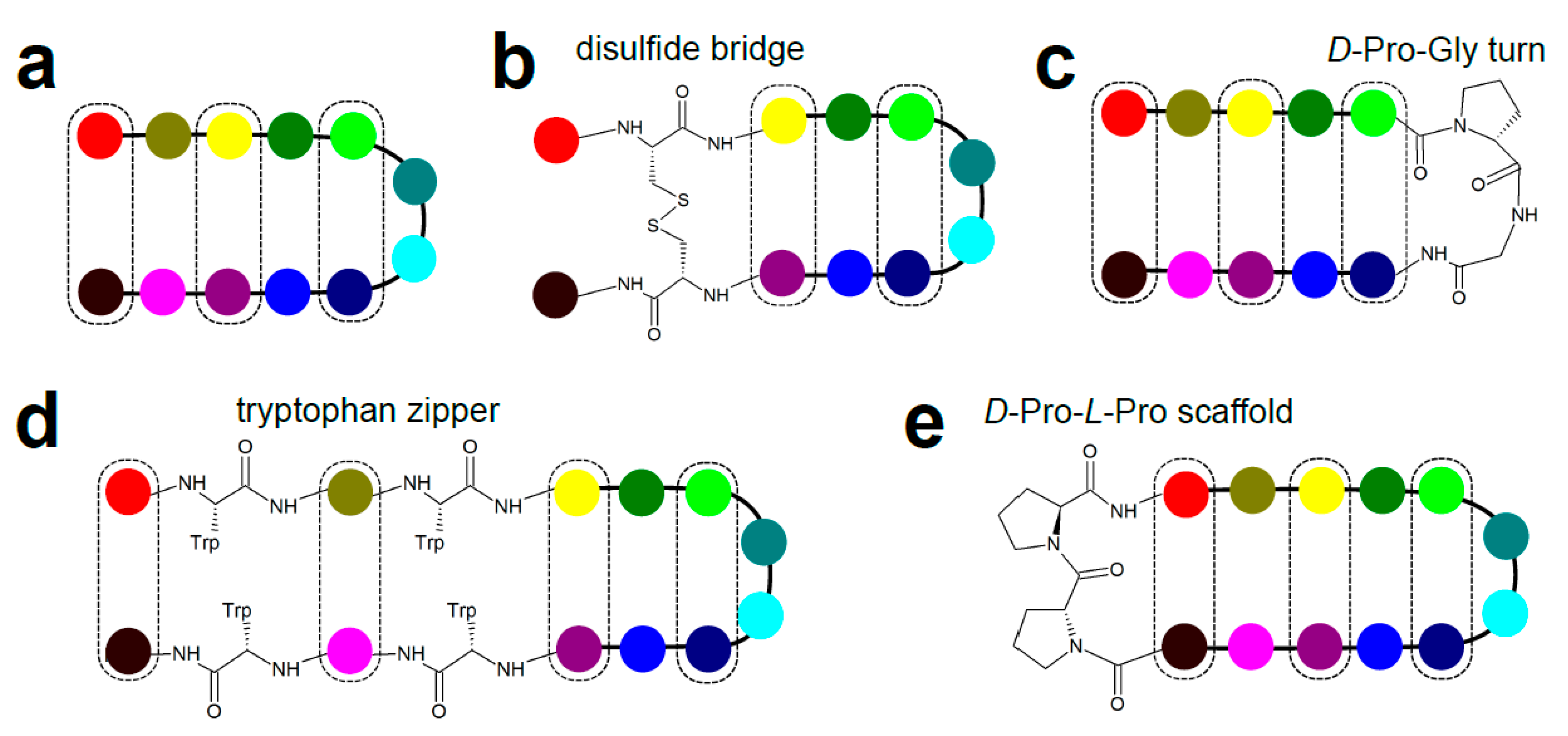
3.5. β-Hairpins and Hairpin Loops
3.6. In Vitro Molecular Evolution
4. Applications of Constrained Peptides
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Empting, M. An introduction to cyclic peptides. In Cyclic Peptides: From Bioorganic Synthesis to Applications; Koehnke, J., Naismith, J., van der Donk, W.A., Eds.; Royal Society of Chemistry: Croydon, UK, 2017; Volume 6, pp. 1–14. [Google Scholar]
- Fang, Z.; Song, Y.; Zhan, P.; Zhang, Q.; Liu, X. Conformational restriction: An effective tactic in ‘follow-on’-based drug discovery. Future Med. Chem. 2014, 6, 885–901. [Google Scholar] [CrossRef]
- Rhodes, C.A.; Pei, D. Bicyclic peptides as next-generation therapeutics. Chemistry 2017, 23, 12690–12703. [Google Scholar] [CrossRef]
- Passioura, T.; Katoh, T.; Goto, Y.; Suga, H. Selection-based discovery of drug-like macrocyclic peptides. Annu. Rev. Biochem. 2014, 83, 727–752. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Domling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Robinson, J.A. Beta-hairpin peptidomimetics: Design, structures and biological activities. Acc. Chem. Res. 2008, 41, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, G.R.; Starovasnik, M.A.; Reynolds, M.E.; Lowman, H.B. A novel family of hairpin peptides that inhibit IgE activity by binding to the high-affinity IgE receptor. Biochemistry 2001, 40, 9828–9835. [Google Scholar] [CrossRef]
- Skelton, N.J.; Russell, S.; de Sauvage, F.; Cochran, A.G. Amino acid determinants of beta-hairpin conformation in erythropoeitin receptor agonist peptides derived from a phage display library. J. Mol. Biol. 2002, 316, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Kasher, R.; Balass, M.; Scherf, T.; Harel, M.; Fridkin, M.; Sussman, J.L.; Fuchs, S. Design and synthesis of peptides that bind alpha-bungarotoxin with high affinity and mimic the three-dimensional structure of the binding-site of acetylcholine receptor. Biophys. Chem. 2003, 100, 293–305. [Google Scholar] [CrossRef]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef]
- Chen, K.; Huang, L.; Shen, B. Rational cyclization-based minimization of entropy penalty upon the binding of Nrf2-derived linear peptides to Keap1: A new strategy to improve therapeutic peptide activity against sepsis. Biophys. Chem. 2019, 244, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Tyndall, J.D.; Fairlie, D.P. Conformational homogeneity in molecular recognition by proteolytic enzymes. J. Mol. Recognit. 1999, 12, 363–370. [Google Scholar] [CrossRef]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 2005, 6, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, D.; Khan, M.M.; Dessen, P.; Held, W.; Huelsken, J.; Heinis, C. Phage selection of peptide macrocycles against beta-catenin to interfere with Wnt signaling. Chem. Med. Chem. 2016, 11, 834–839. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Qian, Z.; Habir, N.A.; Pei, D. Direct Ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron 2014, 70, 7714–7720. [Google Scholar] [CrossRef] [PubMed]
- Adaligil, E.; Patil, K.; Rodenstein, M.; Kumar, K. Discovery of peptide antibiotics composed of D-amino acids. ACS Chem. Biol. 2019, 14, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- ‘t Hart, P.; Wood, T.M.; Tehrani, K.; van Harten, R.M.; Sleszynska, M.; Rentero Rebollo, I.; Hendrickx, A.P.A.; Willems, R.J.L.; Breukink, E.; Martin, N.I. De novo identification of lipid II binding lipopeptides with antibacterial activity against vancomycin-resistant bacteria. Chem. Sci. 2017, 8, 7991–7997. [Google Scholar] [CrossRef]
- Richelle, G.J.J.; Schmidt, M.; Ippel, H.; Hackeng, T.M.; van Maarseveen, J.H.; Nuijens, T.; Timmerman, P. A one-pot “triple-C” multicyclization methodology for the synthesis of highly constrained isomerically pure tetracyclic peptides. Chembiochem 2018, 19, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.B.; Upadhyaya, P.; Qian, Z.; Pei, D. Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb. Sci. 2016, 18, 75–85. [Google Scholar] [CrossRef]
- Rhodes, C.A.; Dougherty, P.G.; Cooper, J.K.; Qian, Z.; Lindert, S.; Wang, Q.E.; Pei, D. Cell-permeable bicyclic peptidyl inhibitors against NEMO-IkappaB kinase interaction directly from a combinatorial library. J. Am. Chem. Soc. 2018, 140, 12102–12110. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-penetrating peptides: Design strategies beyond primary structure and amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Moellering, R.E.; Hilinski, G.J.; Kim, Y.-W.; Grossmann, T.N.; Yehab, J.T.-H.; Verdine, G.L. Towards understanding cell penetration by stapled peptides. Med. Chem. Comm. 2015, 6, 111–119. [Google Scholar] [CrossRef]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Fasan, R.; Dias, R.L.; Moehle, K.; Zerbe, O.; Vrijbloed, J.W.; Obrecht, D.; Robinson, J.A. Using a beta-hairpin to mimic an alpha-helix: Cyclic peptidomimetic inhibitors of the p53-HDM2 protein-protein interaction. Angew. Chem. Int. Ed. Engl. 2004, 43, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.T.; Shortridge, M.D.; Varani, G. A small cyclic beta-hairpin peptide mimics the Rbfox2 RNA recognition motif and binds to the precursor miRNA-20b. Chembiochem 2019, 20, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, M.D.; Walker, M.J.; Pavelitz, T.; Chen, Y.; Yang, W.; Varani, G. A macrocyclic peptide ligand binds the oncogenic microRNA-21 precursor and suppresses dicer processing. ACS Chem. Biol. 2017, 12, 1611–1620. [Google Scholar] [CrossRef]
- Bratkovič, T.; Lunder, M.; Popovič, T.; Kreft, S.; Turk, B.; Štrukelj, B.; Urleb, U. Affinity selection to papain yields potent peptide inhibitors of cathepsins L., B., H., and K. Biochem. Biophys. Res. Commun. 2005, 332, 897–903. [Google Scholar] [CrossRef]
- Bratkovič, T.; Berlec, A.; Popovič, T.; Lunder, M.; Kreft, S.; Urleb, U.; Štrukelj, B. Engineered staphylococcal protein A’s IgG-binding domain with cathepsin L inhibitory activity. Biochem. Biophys. Res. Commun. 2006, 349, 449–453. [Google Scholar] [CrossRef]
- Lu, Z.; Murray, K.S.; Van Cleave, V.; LaVallie, E.R.; Stahl, M.L.; McCoy, J.M. Expression of thioredoxin random peptide libraries on the Escherichia coli cell surface as functional fusions to flagellin: A system designed for exploring protein-protein interactions. Biotechnology 1995, 13, 366–372. [Google Scholar] [CrossRef]
- Borghouts, C.; Kunz, C.; Groner, B. Peptide aptamer libraries. Comb. Chem. High Throughput Screen 2008, 11, 135–145. [Google Scholar]
- Reverdatto, S.; Burz, D.S.; Shekhtman, A. Peptide aptamers: Development and applications. Curr. Top. Med. Chem. 2015, 15, 1082–1101. [Google Scholar] [CrossRef] [PubMed]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef]
- Reguera, L.; Rivera, D.G. Multicomponent reaction toolbox for peptide macrocyclization and stapling. Chem. Rev. 2019, 119, 9836–9860. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Li, Z.; Shao, S.; Ren, X.; Sun, J.; Guo, Z.; Wang, S.; Song, M.M.; Chang, C.A.; Xue, M. Construction of a sequenceable protein mimetic peptide library with a true 3D diversifiable chemical space. J. Am. Chem. Soc. 2018, 140, 14552–14556. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Wong, C.T.T.; Li, X. Chemoselective peptide cyclization and bicyclization directly on unprotected peptides. J. Am. Chem. Soc. 2019, 141, 12274–12279. [Google Scholar] [CrossRef]
- Thapa, P.; Zhang, R.Y.; Menon, V.; Bingham, J.P. Native chemical ligation: A boon to peptide chemistry. Molecules 2014, 19, 14461–14483. [Google Scholar] [CrossRef]
- Malins, L.R.; de Gruyter, J.N.; Robbins, K.J.; Scola, P.M.; Eastgate, M.D.; Ghadiri, M.R.; Baran, P.S. Peptide macrocyclization inspired by non-ribosomal imine natural products. J. Am. Chem. Soc. 2017, 139, 5233–5241. [Google Scholar] [CrossRef]
- Bionda, N.; Cryan, A.L.; Fasan, R. Bioinspired strategy for the ribosomal synthesis of thioether-bridged macrocyclic peptides in bacteria. ACS Chem. Biol. 2014, 9, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Bionda, N.; Fasan, R. Ribosomal synthesis of natural-product-like bicyclic peptides in Escherichia coli. Chembiochem 2015, 16, 2011–2016. [Google Scholar] [CrossRef]
- Scott, C.P.; Abel-Santos, E.; Wall, M.; Wahnon, D.C.; Benkovic, S.J. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Morales-Sanfrutos, J.; Angelini, A.; Cutting, B.; Heinis, C. Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. Chembiochem 2012, 13, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Sindlinger, J.; Schwarzer, D.; Koch, P.; Boeckler, F.M. The symmetric tetravalent sulfhydryl-specific linker NATBA facilitates a combinatorial “tool kit” strategy for phage display-based selection of functionalized bicyclic peptides. ACS Omega 2018, 3, 12361–12368. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.R.; Yu, H.; Wickware, J.M.; Lin, Y.S.; Derda, R. Light-responsive bicyclic peptides. Org. Biomol. Chem 2018, 16, 7588–7594. [Google Scholar] [CrossRef]
- Zou, Y.; Spokoyny, A.M.; Zhang, C.; Simon, M.D.; Yu, H.; Lin, Y.S.; Pentelute, B.L. Convergent diversity-oriented side-chain macrocyclization scan for unprotected polypeptides. Org. Biomol. Chem. 2014, 12, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.S.; Pentelute, B.L. A perfluoroaryl-cysteine SNAr chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef]
- Antos, J.M.; Popp, M.W.; Ernst, R.; Chew, G.L.; Spooner, E.; Ploegh, H.L. A straight path to circular proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, X.; Guo, Z. Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem. Commun. 2011, 47, 9218–9220. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Hemu, X.; Quek, J.P.; Tam, J.P. Butelase-mediated macrocyclization of D-amino-acid-containing peptides. Angew Chem. Int. Ed. Engl. 2016, 55, 12802–12806. [Google Scholar] [CrossRef] [PubMed]
- Braisted, A.C.; Judice, J.K.; Wells, J.A. Synthesis of proteins by subtiligase. Methods Enzymol. 1997, 289, 298–313. [Google Scholar]
- Toplak, A.; Nuijens, T.; Quaedflieg, P.J.L.M.; Wu, B.; Janssen, D.B. Peptiligase, an enzyme for efficient chemoenzymatic peptide synthesis and cyclization in water. Adv. Synth. Catal. 2016, 358, 2140–2147. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.; van Maarseveen, J.H.; Nuijens, T. Enzyme-catalyzed peptide cyclization. Drug Discov. Today Technol. 2017, 26, 11–16. [Google Scholar] [CrossRef]
- Schmidt, M.; Toplak, A.; Quaedflieg, P.J.L.M.; Ippel, H.; Richelle, G.J.J.; Hackeng, T.M.; van Maarseveen, J.H.; Nuijens, T. Omniligase-1: A powerful tool for peptide head-to-tail cyclization. Adv. Synth. Catal. 2017, 359, 2050–2055. [Google Scholar] [CrossRef]
- Schmidt, M.; Huang, Y.H.; Texeira de Oliveira, E.F.; Toplak, A.; Wijma, H.J.; Janssen, D.B.; van Maarseveen, J.H.; Craik, D.J.; Nuijens, T. Efficient enzymatic cyclization of disulfide-rich peptides by using peptide ligases. Chembiochem 2019, 20, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Streefkerk, D.E.; Schmidt, M.; Ippel, J.H.; Hackeng, T.M.; Nuijens, T.; Timmerman, P.; van Maarseveen, J.H. Synthesis of constrained tetracyclic peptides by consecutive CEPS, CLIPS, and oxime ligation. Org. Lett. 2019, 21, 2095–2100. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Grubbs, R.H. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew. Chem. Int. Ed. Engl. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Shepherd, N.E.; Hoang, H.N.; Abbenante, G.; Fairlie, D.P. Single turn peptide alpha helices with exceptional stability in water. J. Am. Chem. Soc. 2005, 127, 2974–2983. [Google Scholar] [CrossRef]
- Scrima, M.; Le Chevalier-Isaad, A.; Rovero, P.; Papini, A.M.; Chorev, M.; D’Ursi, A.M. CuI-catalyzed azide-alkyne intramolecular i-to-(i+4) side-chain-to-side-chain cyclization promotes the formation of helix-like secondary structures. Eur. J. Org. Chem. 2010, 3, 446–457. [Google Scholar] [CrossRef]
- Madden, M.M.; Rivera Vera, C.I.; Song, W.; Lin, Q. Facile synthesis of stapled, structurally reinforced peptide helices via a photoinduced intramolecular 1,3-dipolar cycloaddition reaction. Chem. Commun. 2009, 37, 5588–5590. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; King, D.S.; Chmielewski, J.; Singh, S.; Schultz, P.G. General approach to the synthesis of short alpha-helical peptides. J. Am. Chem. Soc. 1991, 113, 9391–9392. [Google Scholar] [CrossRef]
- Brunel, F.M.; Dawson, P.E. Synthesis of constrained helical peptides by thioether ligation: Application to analogs of gp41. Chem. Commun. 2005, 20, 2552–2554. [Google Scholar] [CrossRef] [PubMed]
- Assem, N.; Ferreira, D.J.; Wolan, D.W.; Dawson, P.E. Acetone-linked peptides: A convergent approach for peptide macrocyclization and labeling. Angew. Chem. Int. Ed. Engl. 2015, 54, 8665–8668. [Google Scholar] [CrossRef]
- Hilinski, G.J.; Kim, Y.W.; Hong, J.; Kutchukian, P.S.; Crenshaw, C.M.; Berkovitch, S.S.; Chang, A.; Ham, S.; Verdine, G.L. Stitched alpha-helical peptides via bis ring-closing metathesis. J. Am. Chem. Soc. 2014, 136, 12314–12322. [Google Scholar] [CrossRef]
- Serrano, J.C.; Sipthorp, J.; Xu, W.; Itzhaki, L.S.; Ley, S.V. A new methodology for incorporating chiral linkers into stapled peptides. Chembiochem 2017, 18, 1066–1071. [Google Scholar] [CrossRef]
- Hu, K.; Geng, H.; Zhang, Q.; Liu, Q.; Xie, M.; Sun, C.; Li, W.; Lin, H.; Jiang, F.; Wang, T.; et al. An in-tether chiral center modulates the helicity, cell permeability, and target binding affinity of a peptide. Angew. Chem. Int. Ed. Engl. 2016, 55, 8013–8017. [Google Scholar] [CrossRef]
- Jiang, Y.; Hu, K.; Shi, X.; Tang, Q.; Wang, Z.; Ye, X.; Li, Z. Switching substitution groups on the in-tether chiral centre influences backbone peptides’ permeability and target binding affinity. Org. Biomol. Chem. 2017, 15, 541544. [Google Scholar] [CrossRef]
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. [Google Scholar] [CrossRef]
- Obrecht, D.; Chevalier, E.; Moehle, K.; Robinson, J.A. Beta-hairpin protein epitope mimetic technology in drug discovery. Drug Discov. Today Technol. 2012, 9, e1–e70. [Google Scholar] [CrossRef]
- Cochran, A.G.; Tong, R.T.; Starovasnik, M.A.; Park, E.J.; McDowell, R.S.; Theaker, J.E.; Skelton, N.J. A minimal peptide scaffold for beta-turn display: Optimizing a strand position in disulfide-cyclized beta-hairpins. J. Am. Chem. Soc. 2001, 123, 625–632. [Google Scholar] [CrossRef]
- Cochran, A.G.; Skelton, N.J.; Starovasnik, M.A. Tryptophan zippers: Stable, monomeric beta -hairpins. Proc. Natl. Acad. Sci. USA 2001, 98, 5578–5583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Campbell, R.E. An engineered tryptophan zipper-type peptide as a molecular recognition scaffold. J. Pept. Sci. 2009, 15, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M.; Blanco, F.J.; Serrano, L. De novo design and structural analysis of a model beta-hairpin peptide system. Nat. Struct. Biol. 1996, 3, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Stanger, H.E.; Gellman, S.H. Rules for antiparallel β-sheet design: D-Pro-Gly is superior to L-Asn-Gly for β-hairpin nucleation. J. Am. Chem. Soc. 1998, 120, 4236–4237. [Google Scholar] [CrossRef]
- Masterson, L.R.; Etienne, M.A.; Porcelli, F.; Barany, G.; Hammer, R.P.; Veglia, G. Nonstereogenic alpha-aminoisobutyryl-glycyl dipeptidyl unit nucleates type I’ beta-turn in linear peptides in aqueous solution. Biopolymers 2007, 88, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Raghothama, S.; Sridharan, R.; Balaram, P. Tuning the beta-turn segment in designed peptide beta-hairpins: Construction of a stable type I’ beta-turn nucleus and hairpin-helix transition promoting segments. Biopolymers 2007, 88, 350–361. [Google Scholar] [CrossRef]
- Raghavender, U.S.; Aravinda, S.; Rai, R.; Shamala, N.; Balaram, P. Peptide hairpin nucleation with the obligatory Type I’ beta-turn Aib-DPro segment. Org. Biomol. Chem. 2010, 8, 3133–3135. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Moehle, K.; Jiang, L.; Pfeiffer, B.; Robinson, J.A. Structural mimicry of canonical conformations in antibody hypervariable loops using cyclic peptides containing a heterochiral diproline template. J. Am. Chem. Soc. 1999, 121, 2679–2685. [Google Scholar] [CrossRef]
- Descours, A.; Moehle, K.; Renard, A.; Robinson, J.A. A new family of beta-hairpin mimetics based on a trypsin inhibitor from sunflower seeds. Chembiochem 2002, 3, 318–323. [Google Scholar] [CrossRef]
- Dias, R.L.; Fasan, R.; Moehle, K.; Renard, A.; Obrecht, D.; Robinson, J.A. Protein ligand design: From phage display to synthetic protein epitope mimetics in human antibody Fc-binding peptidomimetics. J. Am. Chem. Soc. 2006, 128, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Ishiguro, K.; Takahashi, T.; Mihara, H. A novel beta-loop scaffold of phage-displayed peptides for highly specific affinities. Mol. Biosyst. 2011, 7, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.T.; Lopez de la Paz, M.; Lacroix, E.; Serrano, L.; Perez-Paya, E. Combinatorial approaches: A new tool to search for highly structured beta-hairpin peptides. Proc. Natl. Acad. Sci. USA 2002, 99, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Mulligan, V.K.; Bahl, C.D.; Gilmore, J.M.; Harvey, P.J.; Cheneval, O.; Buchko, G.W.; Pulavarti, S.V.; Kaas, Q.; Eletsky, A.; et al. Accurate de novo design of hyperstable constrained peptides. Nature 2016, 538, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Bozovičar, K.; Bratkovič, T. Evolving a peptide: Library platforms and diversification strategies. Int. J. Mol. Sci. 2020, 21, 215. [Google Scholar] [CrossRef]
- Rentero Rebollo, I.; Heinis, C. Phage selection of bicyclic peptides. Methods 2013, 60, 46–54. [Google Scholar] [CrossRef]
- Chen, S.; Rentero Rebollo, I.; Buth, S.A.; Morales-Sanfrutos, J.; Touati, J.; Leiman, P.G.; Heinis, C. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J. Am. Chem. Soc. 2013, 135, 6562–6569. [Google Scholar] [CrossRef]
- Chen, S.; Heinis, C. Phage selection of bicyclic peptides based on two disulfide bridges. Methods Mol. Biol. 2015, 1248, 119–137. [Google Scholar]
- Guillen Schlippe, Y.V.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In vitro selection of highly modified cyclic peptides that act as tight binding inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar] [CrossRef] [PubMed]
- Cervettini, D.; Tang, S.; Fried, S.D.; Willis, J.C.W.; Funke, L.F.H.; Colwell, L.J.; Chin, J.W. Rapid discovery and evolution of orthogonal aminoacyl-tRNA synthetase-tRNA pairs. Nat. Biotechnol. 2020, 38, 989–999. [Google Scholar] [CrossRef]
- Iwasaki, K.; Goto, Y.; Katoh, T.; Suga, H. Selective thioether macrocyclization of peptides having the N-terminal 2-chloroacetyl group and competing two or three cysteine residues in translation. Org. Biomol. Chem. 2012, 10, 5783–5786. [Google Scholar] [CrossRef] [PubMed]
- Okuma, R.; Kuwahara, T.; Yoshikane, T.; Watanabe, M.; Dranchak, P.; Inglese, J.; Shuto, S.; Goto, Y.; Suga, H. A macrocyclic peptide library with a structurally constrained cyclopropane-containing building block leads to thiol-independent inhibitors of phosphoglycerate mutase. Chem. Asian J. 2020, 15, 2631–2636. [Google Scholar] [CrossRef]
- Gonzalez-Navarro, H.; Mora, P.; Pastor, M.; Serrano, L.; Mingarro, I.; Perez-Paya, E. Identification of peptides that neutralize bacterial endotoxins using beta-hairpin conformationally restricted libraries. Mol. Divers 2000, 5, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, Z.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in P. aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef]
- Timmerman, P.; Puijk, W.C.; Meloen, R.H. Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J. Mol. Recognit. 2007, 20, 283–299. [Google Scholar] [CrossRef]
- Iqbal, E.S.; Richardson, S.L.; Abrigo, N.A.; Dods, K.K.; Osorio Franco, H.E.; Gerrish, H.S.; Kotapati, H.K.; Morgan, I.M.; Masterson, D.S.; Hartman, M.C.T. A new strategy for the in vitro selection of stapled peptide inhibitors by mRNA display. Chem. Commun. (Camb) 2019, 55, 8959–8962. [Google Scholar] [CrossRef]
- Anananuchatkul, T.; Tsutsumi, H.; Miki, T.; Mihara, H. hDM2 protein-binding peptides screened from stapled alpha-helical peptide phage display libraries with different types of staple linkers. Bioorg. Med. Chem. Lett. 2020, 30, 127605. [Google Scholar] [CrossRef] [PubMed]
- Anananuchatkul, T.; Chang, I.V.; Miki, T.; Tsutsumi, H.; Mihara, H. Construction of a stapled alpha-helix peptide library displayed on phage for the screening of galectin-3-binding peptide ligands. ACS Omega 2020, 5, 5666–5674. [Google Scholar] [CrossRef]
- Timmerman, P.; Barderas, R.; Desmet, J.; Altschuh, D.; Shochat, S.; Hollestelle, M.J.; Höppener, J.W.M.; Monasterio, A.; Ignacio Casal, J.; Meloen, R.H. A combinatorial approach for the design of complementarity-determining region-derived peptidomimetics with in vitro anti-tumoral activity. J. Biol. Chem. 2009, 284, 34126–34134. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE delivery using the bicycle toxin conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Gowland, C.; Berry, P.; Errington, J.; Jeffrey, P.; Bennett, G.; Godfrey, L.; Pittman, M.; Niewiarowski, A.; Symeonides, N.S.; Veal, G.J. Development of a LC-MS/MS method for the quantification of toxic payload DM1 cleaved from BT1718 in a phase I study. Bioanalysis 2021, 13, 101–113. [Google Scholar] [CrossRef]
- Bicycle Therapeutics Programs. Available online: https://www.bicycletherapeutics.com/programs (accessed on 29 January 2021).
- Bernhagen, D.; Jungbluth, V.; Gisbert Quilis, N.; Dostalek, J.; White, P.B.; Jalink, K.; Timmerman, P. High-affinity alpha5beta1-integrin-selective bicyclic RGD peptides identified via screening of designed random libraries. ACS Comb. Sci. 2019, 21, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, D.; Jungbluth, V.; Quilis, N.G.; Dostalek, J.; White, P.B.; Jalink, K.; Timmerman, P. Bicyclic RGD peptides with exquisite selectivity for the integrin alphavbeta3 receptor using a “random design” approach. ACS Comb. Sci. 2019, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Tamada, K.; Nakajima, S.; Ogawa, N.; Inada, M.; Shibasaki, H.; Sato, A.; Takasawa, R.; Yoshimori, A.; Suzuki, Y.; Watanabe, N.; et al. Papaverine identified as an inhibitor of high mobility group box 1/receptor for advanced glycation end-products interaction suppresses high mobility group box 1-mediated inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 511, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Sclavons, C.; Burtea, C.; Boutry, S.; Laurent, S.; Vander Elst, L.; Muller, R.N. Phage display screening for tumor necrosis factor-alpha-binding peptides: Detection of inflammation in a mouse model of hepatitis. Int. J. Pept. 2013, 2013, 348409. [Google Scholar] [CrossRef]
- Bezer, S.; Matsumoto, M.; Lodewyk, M.W.; Lee, S.J.; Tantillo, D.J.; Gagne, M.R.; Waters, M.L. Identification and optimization of short helical peptides with novel reactive functionality as catalysts for acyl transfer by reactive tagging. Org. Biomol. Chem. 2014, 12, 1488–1494. [Google Scholar] [CrossRef]
- Matsumoto, M.; Lee, S.J.; Gagne, M.R.; Waters, M.L. Cross-strand histidine-aromatic interactions enhance acyl-transfer rates in beta-hairpin peptide catalysts. Org. Biomol. Chem. 2014, 12, 8711–8718. [Google Scholar] [CrossRef]
- Matsumoto, M.; Lee, S.J.; Waters, M.L.; Gagne, M.R. A catalyst selection protocol that identifies biomimetic motifs from beta-hairpin libraries. J. Am. Chem. Soc. 2014, 136, 15817–15820. [Google Scholar] [CrossRef]
- Cary, D.R.; Ohuchi, M.; Reid, P.C.; Masuya, K. Constrained peptides in drug discovery and development. J. Synth. Organ. Chem. Jpn. 2017, 75, 1171–1178. [Google Scholar] [CrossRef]
- Shenderovich, M.D.; Liao, S.; Qian, X.; Hruby, V.J. A three-dimensional model of the delta-opioid pharmacophore: Comparative molecular modeling of peptide and nonpeptide ligands. Biopolymers 2000, 53, 565–580. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Kouznetsova, V.L.; Biswas, N.; Mahata, S.K.; O’Connor, D.T. Development of a pharmacophore model for the catecholamine release-inhibitory peptide catestatin: Virtual screening and functional testing identify novel small molecule therapeutics of hypertension. Bioorg. Med. Chem. 2013, 21, 5855–5869. [Google Scholar] [CrossRef] [PubMed]











| Chemistry | Residues Involved | Compatible Arrangement | Does Stereochemistry of the Staple Handles Need to be Considered? | Comment |
|---|---|---|---|---|
| alkene ring-closing metathesis | (homo)serine O-allyl ethers [59] | [i, i + 4] | no | all hydrocarbon staple |
| α,α-disubstituted residues with olefinic side chains (R or S configuration, 5 or 8 atoms long) [60] | [i, i + 4] and [i, i + 7] | yes (S5/S5 for [i, i + 4], S8/R5 or S5/R8 for [i, i + 7]) | ||
| lactamisation | lysine and glutamate, or ornithine and aspartate [61] | only compatible with [i, i + 4] arrangement | no | requires extra orthogonal protective groups for amino and carboxy groups for on resin lactamisation |
| cycloadditions | azide and alkyne group containing residues with 4 + 2 or 4 + 3 methylene units long side chains [62] | [i, i + 4] | no | well-established click reaction (Cu(I)-catalyzed azide-alkyne cycloaddition) |
| tetrazole and alkene group containing residues [63] | UV-induced cycloaddition between tetrazoles and alkenes to yield fluorescent pyrazoline tethers | |||
| disulfide bridges | thiol group containing residues [64] | [i, i + 7] | yes (combination of D and L-residues) | chronologically the oldest technique, requires acetamidomethyl protecting groups for thiols, staple unstable (prone to reduction) |
| thioether bridges | cysteine and an alpha-bromo amide group containing residue [65] | [i, i + 3] and [i, i + 4] | no | staple stable, higher helicity achieved with [i, i + 3] arrangement |
| two (homo)cysteines + dichloroacetone crosslinker [66] | [i, i + 4] | no | bis-alkylating crosslinker amenable to further derivation via oxime ligation (e.g., fluorophore or biotin coupling) | |
| cysteines + perfluoroaromatic crosslinker (e.g., hexafluorobenzene) [48] | [i, i + 4] | no | regioselective reaction (para-disubstituted staple) proceeding under mild conditions in high yield even for unprotected peptides |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozovičar, K.; Bratkovič, T. Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. Int. J. Mol. Sci. 2021, 22, 1611. https://doi.org/10.3390/ijms22041611
Bozovičar K, Bratkovič T. Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. International Journal of Molecular Sciences. 2021; 22(4):1611. https://doi.org/10.3390/ijms22041611
Chicago/Turabian StyleBozovičar, Krištof, and Tomaž Bratkovič. 2021. "Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties" International Journal of Molecular Sciences 22, no. 4: 1611. https://doi.org/10.3390/ijms22041611
APA StyleBozovičar, K., & Bratkovič, T. (2021). Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. International Journal of Molecular Sciences, 22(4), 1611. https://doi.org/10.3390/ijms22041611