
The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia


Abstract
1. Introduction
2. Criteria of Cancer Cachexia
3. Cancer Cachexia and Systemic Inflammatory Response (SIR)
3.1. Tumor Necrosis Factor α (TNF-α) and Proteolysis-Inducing Factor (PIF)
3.2. Interleukin 1 α and β (IL-1α, IL-1β)
3.3. Interleukin 6 (IL-6)
4. Systemic Inflammatory Response in Colorectal Cancer-Associated Cachexia
5. The Role of Tumor Microenvironment (TME) Cells in CRC-Associated Cachexia
5.1. Tumor-Infiltrating Immune Cells (TIICs)
5.1.1. Tumor-Associated Macrophages (TAMs)
5.1.2. Tumor Infiltrating Lymphocytes (TILs)
5.1.3. Tumor-Associated Neutrophils (TANs)
5.1.4. Myeloid-Derived Suppressor Cells (MDSCs)
5.2. Cancer-Associated Adipocytes (CAAs), Tumor-Resident Adipocytes (TRAs), Adipose Stromal Cells (ASCs)
5.3. Cancer-Associated Fibroblasts (CAFs)
5.4. Colorectal Cancer Cells
6. The Main Signaling Pathways Involved in Skeletal-Muscle and Adipose-Tissue Alterations in Cancer Cachexia
6.1. Skeletal Muscle
6.2. Adipose Tissue
7. Autophagy and Cancer Cachexia—Role of TME Cells
8. Therapeutical Options for Cancer Cachexia in CRC
9. Concluding Remarks and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAs | Amino Acids |
| ACC | Acetyl-CoA Carboxylase |
| ADAMs | A Disintegrin and Metalloprotease |
| Akt | Serine/threonine protein kinase |
| AMPK | AMP-activated Protein Kinase |
| APC | Adenomatous Polyposis Coli |
| AT/WAT | Adipose Tissue/White AT |
| ATGL | Adipose Triglyceride Lipase |
| CAAs | Cancer-associated Adipocytes |
| CAFs | Cancer-associated Fibroblasts |
| C26 | Colon Adenocarcinoma 26 Cells |
| CC | Cancer Cachexia |
| CCL2/MCP-1 | Chemokine (C-C Motif) Ligand 2/Monocyte Chemoattractant Protein 1 |
| CCL3/MIP-1α | Chemokine (C-C Motif) Ligand 3/Macrophage Inflammatory Protein-1 α |
| CD3, -4, -8 | Cluster of Differentiation 3, -4, -8 |
| CIFs | Cachexia-inducing Factors |
| COX-2 | Cyclooxygenase-2 |
| CPT-1 | Carnitine Palmitoyltransferase 1 |
| CRC | Colorectal Cancer |
| CRP | C-reactive Protein |
| CSS | Cancer-specific Survival |
| CXCL8/IL-8 | Chemokine (C-X-C Motif) Ligand 8/Interleukin 8 |
| DCs | Dendritic Cells |
| DFS | Disease Free Survival |
| ECs | Endothelial Cells |
| ECM | Extracellular Matrix |
| EGF | Epidermal Growth Factor |
| eIF2 | Eukaryotic Initiation Factor 2 |
| EMT | Epithelial-Mesenchymal Transition |
| ERK | Extracellular-regulated Kinase |
| FADD | FAS-associated Death Domain |
| FFA | Free Fatty Acid |
| Fn14 | Receptor of the TWEAK |
| FOXQ1 | Forkhead Box Q1 Protein |
| FSP1 | Fibroblast-specific Protein 1 |
| FSTL1 | Follistatin-related Protein 1 |
| G(M)-CSF | Granulocyte(Macrophage)-Colony-stimulating Factor |
| GPIHBP1 | Glycosylphosphatidylinositol-anchored High-density Lipoprotein Binding Protein 1 |
| HIF-1α | Hypoxia-inducible Factor 1 alpha |
| HMGB1 | High Mobility Group Box 1 |
| HSL | Hormone-sensitive Lipase |
| IFN-α, β, γ | Interferon alpha, beta, gamma |
| IGF1 | Insulin-like Growth Factor 1 |
| s(IL-)2R, IL-1R1 | Soluble (Interleukin) 2 Receptor; IL-1Receptor type I |
| IP-10 | Interferon gamma-induced protein 10/CXCL10 |
| JAK2 | Janus Kinase 2 |
| LIF | Leukemia Inhibitory Factor |
| LMR | Lymphocyte:Monocyte Ratio |
| mABs | Monoclonal Antibody |
| MAFbx | Muscle Atrophy F-box |
| MAPK | Mitogen-activated Protein Kinase |
| MDSCs | Myeloid-derived Suppressor Cells |
| MEK1/2 | Mitogen-activated Protein Kinase (MAP2K, MEK, MAPKK) |
| mGPS | Modified Glasgow Prognostic Score |
| MLNs | Mesenteric Lymph Nodes |
| MSI-H | High Microsatellite Instability |
| MSS | Microsatellite Stability |
| mTORC1 | Mammalian Target of Rapamycin Complex 1 |
| MuRF1 | Muscle RING-finger 1 |
| MyoD | Myoblast Determination Protein 1 |
| NF-κB | Nuclear Factor kappa B |
| NK | Natural Killer Cells |
| NLR | Neutrophil:Lymphocyte Ratio |
| NOS2 | Nitric Oxidase Synthase 2 |
| OS | Overall Survival |
| PBMCs | Peripheral Blood Mononuclear Cells |
| PD-L1 | Programmed Death-Ligand 1 |
| PGE2/EP2 | Prostaglandin E2 Receptor 2 Subtype |
| PIF | Proteolysis-inducing Factor |
| PI3K | Phosphatidylinositol 3 Kinase |
| PP | Peyer’s Patches |
| RAGE | Receptor for Advanced Glycation End-products |
| RANTES | Regulated on Activation, Normal T-cell Expressed and Secreted |
| RFS | Recurrence-free Survival |
| ROS | Reactive Oxygen Species |
| SDS-MYL1 | Sodium Dodecyl Sulfate Soluble Myosin Light Chain 1 |
| SII | Systemic Inflammatory Index |
| SIR | Systemic Inflammatory Response |
| SMI | Skeletal-Muscle Index |
| SOCS3 | Suppressor of Cytokine Signaling 3 |
| SSAT | Spermidine/Spermine N-1 Acetyl Transferase |
| STAT1, -3 | Signal Transducer and Activator of Transcription Protein 1, 3 |
| TAMs | Tumor-associated Macrophages |
| TANs | Tumor-associated Neutrophils |
| TGF-β | Tumor Growth Factor beta |
| TIICs | Tumor-infiltrating Immune Cells |
| TILs | Tumor-infiltrating Lymphocytes |
| TME | Tumor Microenvironment |
| TNF-α, TNFR2 | Tumor Necrosis Factor α/TNF Receptor 2 |
| Treg | Regulatory T Cells |
| TWEAK | TNF-like Weak Inducer of Apoptosis |
| UPC1 | Uncoupling Protein 1 |
| UPS | Ubiquitin-proteasome System |
| VEGF-A | Vascular Endothelial Growth Factor A |
| WBC | White Blood Cells |
| Wnt | Wnt (gene wingless + integrated or int-1) Family Member |
| ZAG | Zinc-α2-Glycoprotein |
References
- Vagnildhaug, O.M.; Balstad, T.R.; Almberg, S.S.; Brunelli, C.; Knudsen, A.K.; Kaasa, S.; Thronæs, M.; Laird, B.; Solheim, T.S. A cross-sectional study examining the prevalence of cachexia and areas of unmet need in patients with cancer. Support Care Cancer 2018, 26, 1871–1880. [Google Scholar] [CrossRef]
- Dev, R. Measuring cachexia-diagnostic criteria. Ann. Palliat. Med. 2019, 8, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kumar, A. ER Stress and Unfolded Protein Response in Cancer Cachexia. Cancers 2019, 11, 1929. [Google Scholar] [CrossRef] [PubMed]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.J.; Patel, B.M. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Novinger, L.J.; Pin, F.; Bonetto, A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis. Model. Mech. 2020, 13, dmm043166. [Google Scholar] [CrossRef]
- Peixoto da Silva, S.; Santos, J.M.O.; Costa, E.; Silva, M.P.; Gil da Costa, R.M.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachexia Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef]
- Shum, A.M.; Fung, D.C.; Corley, S.M.; McGill, M.C.; Bentley, N.L.; Tan, T.C.; Wilkins, M.R.; Polly, P. Cardiac and skeletal muscles show molecularly distinct responses to cancer cachexia. Physiol. Genom. 2015, 47, 588–599. [Google Scholar] [CrossRef]
- Belloum, Y.; Rannou-Bekono, F.; Favier, F.B. Cancer-induced cardiac cachexia: Pathogenesis and impact of physical activity (Review). Oncol. Rep. 2017, 37, 2543–2552. [Google Scholar] [CrossRef]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell. Physiol. 2018, 234, 13–22. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Costa, R.G.F.; Caro, P.L.; de Matos-Neto, E.M.; Lima, J.D.C.C.; Radloff, K.; Alves, M.J.; Camargo, R.G.; Pessoa, A.F.M.; Simoes, E.; Gama, P.; et al. Cancer cachexia induces morphological and inflammatory changes in the intestinal mucosa. J. Cachexia Sarcopenia Muscle 2019, 10, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Anker, M.S.; Holcomb, R.; Muscaritoli, M.; von Haehling, S.; Haverkamp, W.; Jatoi, A.; Morley, J.E.; Strasser, F.; Landmesser, U.; Coats, A.J.S.; et al. Orphan disease status of cancer cachexia in the USA and in the European Union: A systematic review. J. Cachexia Sarcopenia Muscle 2019, 10, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Zhang, L. Cancer Cachexia: Definition, Staging, and Emerging Treatments. Cancer Manag. Res. 2020, 12, 5597–5605. [Google Scholar] [CrossRef]
- Malietzis, G.; Currie, A.C.; Athanasiou, T.; Johns, N.; Anyamene, N.; Glynne-Jones, R.; Kennedy, R.H.; Fearon, K.C.; Jenkins, J.T. Influence of body composition profile on outcomes following colorectal cancer surgery. Br. J. Surg. 2016, 103, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Sueda, T.; Takahasi, H.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Doki, Y.; Mori, M. Impact of Low Muscularity and Myosteatosis on Long-term Outcome After Curative Colorectal Cancer Surgery: A Propensity Score-Matched Analysis. Dis. Colon Rectum 2018, 61, 364–374. [Google Scholar] [CrossRef]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Tabuso, M.; Homer-Vanniasinkam, S.; Adya, R.; Arasaradnam, R.P. Role of tissue microenvironment resident adipocytes in colon cancer. World J. Gastroenterol. 2017, 23, 5829–5835. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Chiba, F.; Soda, K.; Yamada, S.; Tokutake, Y.; Chohnan, S.; Konishi, F.; Rikiyama, T. The importance of tissue environment surrounding the tumor on the development of cancer cachexia. Int. J. Oncol. 2014, 44, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Al-Zoughbi, W.; Hoefler, G. Tumor Macroenvironment: An Update. Pathobiology 2020, 87, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S.M.; Pitteri, S.J.; Faca, V.M. Mining the plasma proteome for cancer biomarkers. Nature 2008, 452, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Inácio Pinto, N.; Carnier, J.; Oyama, L.M.; Otoch, J.P.; Alcântara, P.S.; Tokeshi, F.; Nascimento, C.M. Cancer as a Pro-inflammatory Environment: Metastasis and Cachexia. Mediat. Inflamm. 2015, 2015, 791060. [Google Scholar] [CrossRef] [PubMed]
- Freire, P.P.; Fernandez, G.J.; de Moraes, D.; Cury, S.S.; Dal Pai-Silva, M.; Dos Reis, P.P.; Rogatto, S.R.; Carvalho, R.F. The expression landscape of cachexia-inducing factors in human cancers. J. Cachexia Sarcopenia Muscle 2020, 11, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.J.; Figuerêdo, R.G.; Azevedo, F.F.; Cavallaro, D.A.; Neto, N.I.; Lima, J.D.; Matos-Neto, E.; Radloff, K.; Riccardi, D.M.; Camargo, R.G.; et al. Adipose tissue fibrosis in human cancer cachexia: The role of TGFβ pathway. BMC Cancer 2017, 17, 190. [Google Scholar] [CrossRef]
- Lima, J.D.C.C.; Simoes, E.; de Castro, G.; Morais, M.R.P.T.; de Matos-Neto, E.M.; Alves, M.J.; Pinto, N.I.; Figueredo, R.G.; Zorn, T.M.T.; Felipe-Silva, A.S.; et al. Tumour-derived transforming growth factor-β signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1045–1059. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, J.; Zhao, Z.; Yang, M.; Chen, M.; Liu, C.; Ji, J.; Zhu, D. Single-cell transcriptome analysis reveals tumor immune microenvironment heterogenicity and granulocytes enrichment in colorectal cancer liver metastases. Cancer Lett. 2020, 470, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Koi, M.; Carethers, J.M. The colorectal cancer immune microenvironment and approach to immunotherapies. Future Oncol. 2017, 13, 1633–1647. [Google Scholar] [CrossRef] [PubMed]
- Bozzetti, F.; Mariani, L. Defining and classifying cancer cachexia: A proposal by the SCRINIO Working Group. JPEN J. Parenter. Enter. Nutr. 2009, 33, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, B.; Liu, H.; Yang, K.; Thapa, S.; Zhang, H.; Li, L.; Yu, S. Development and validation of a clinically applicable score to classify cachexia stages in advanced cancer patients. J. Cachexia Sarcopenia Muscle 2018, 9, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, C.; Jouinot, A.; Durand, J.P.; Neveux, N.; Boudou-Rouquette, P.; Huillard, O.; Alexandre, J.; Cynober, L.; Goldwasser, F. Relation between hypermetabolism, cachexia, and survival in cancer patients: A prospective study in 390 cancer patients before initiation of anticancer therapy. Am. J. Clin. Nutr. 2017, 105, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, L.; Frykholm, G.; Lydersen, S.; Ulveland, H.; Baracos, V.; Prado, C.M.; Birdsell, L.; Falkmer, U. Nutritional status, cachexia and survival in patients with advanced colorectal carcinoma. Different assessment criteria for nutritional status provide unequal results. Clin. Nutr. 2013, 32, 65–72. [Google Scholar] [CrossRef]
- Lopes, J.P.; Pereira, P.M.D.C.C.; Vicente, A.F.D.R.B.; Bernardo, A.; de Mesquita, M.F. Nutritional status assessment in colorectal cancer patients. Nutr. Hosp. 2013, 28, 412–418. [Google Scholar] [CrossRef]
- Ziętarska, M.; Krawczyk-Lipiec, J.; Kraj, L.; Zaucha, R.; Małgorzewicz, S. Nutritional status assessment in colorectal cancer patients qualified to systemic treatment. Contemp. Oncol. 2017, 21, 157–161. [Google Scholar] [CrossRef]
- Okugawa, Y.; Toiyama, Y.; Yamamoto, A.; Shigemori, T.; Kitamura, A.; Ichikawa, T.; Ide, S.; Kitajima, T.; Fujikawa, H.; Yasuda, H.; et al. Close Relationship Between Immunological/Inflammatory Markers and Myopenia and Myosteatosis in Patients with Colorectal Cancer: A Propensity Score Matching Analysis. JPEN J. Parenter. Enter. Nutr. 2019, 43, 508–515. [Google Scholar] [CrossRef]
- Tisdale, M.J. Catabolic mediators of cancer cachexia. Curr. Opin. Support Palliat. Care 2008, 2, 256–261. [Google Scholar] [CrossRef]
- Onesti, J.K.; Guttridge, D.C. Inflammation based regulation of cancer cachexia. BioMed Res. Int. 2014, 2014, 168407. [Google Scholar] [CrossRef]
- Beutler, B.; Cerami, A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature 1986, 320, 584–588. [Google Scholar] [CrossRef]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef]
- Mirza, K.A.; Tisdale, M.J. Role of Ca2+ in proteolysis-inducing factor (PIF)-induced atrophy of skeletal muscle. Cell. Signal. 2012, 24, 2118–2122. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.J.; McMillan, D.C.; Laird, B.J.A. Targeting IL-1α in cancer cachexia: A narrative review. Curr. Opin. Support Palliat. Care 2018, 12, 453–459. [Google Scholar] [CrossRef]
- Zhang, W.; Borcherding, N.; Kolb, R. IL-1 Signaling in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Apte, R.N. Targeting the Tumor Microenvironment by Intervention in Interleukin-1 Biology. Curr. Pharm. Des. 2017, 23, 4893–4905. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Voronov, E. Immunotherapeutic approaches of IL-1 neutralization in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef]
- Yamashita, A.S.; das Neves, R.X.; Rosa-Neto, J.C.; Lira, F.D.; Batista, M.L., Jr.; Alcantara, P.S.; Otoch, J.P.; Seelaender, M. White adipose tissue IFN-γ expression and signalling along the progression of rodent cancer cachexia. Cytokine 2017, 89, 122–126. [Google Scholar] [CrossRef]
- Webster, J.M.; Kempen, L.J.A.P.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef]
- Bonetto, A.; Rupert, J.E.; Barreto, R.; Zimmers, T.A. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp. 2016, 117, 54893. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cánoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Velázquez, K.T.; Baltgalvis, K.A.; Sato, S.; Baynes, J.W.; Carson, J.A. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J. Cachexia Sarcopenia Muscle 2012, 3, 117–137. [Google Scholar] [CrossRef]
- Hardee, J.P.; Montalvo, R.N.; Carson, J.A. Linking Cancer Cachexia-Induced Anabolic Resistance to Skeletal Muscle Oxidative Metabolism. Oxid. Med. Cell. Longev. 2017, 2017, 8018197. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.M.D.R.; das Neves, R.X.; de Matos-Neto, E.M.; Camargo, R.G.; Lima, J.D.C.C.; Radloff, K.; Alves, M.J.; Costa, R.G.F.; Tokeshi, F.; Otoch, J.P.; et al. Plasma Lipid Profile and Systemic Inflammation in Patients with Cancer Cachexia. Front. Nutr. 2020, 7, 4. [Google Scholar] [CrossRef]
- Scheede-Bergdahl, C.; Watt, H.L.; Trutschnigg, B.; Kilgour, R.D.; Haggarty, A.; Lucar, E.; Vigano, A. Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin. Nutr. 2012, 31, 85–88. [Google Scholar] [CrossRef]
- Talbert, E.E.; Lewis, H.L.; Farren, M.R.; Ramsey, M.L.; Chakedis, J.M.; Rajasekera, P.; Haverick, E.; Sarna, A.; Bloomston, M.; Pawlik, T.M.; et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naïve pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 2018, 9, 358–368. [Google Scholar] [CrossRef]
- Cao, Z.; Jose, I.; Glab, J.; Puthalakath, H.; Osellame, L.D.; Hoogenraad, N.J. Generation of reporter cell lines for factors inducing muscle wasting in cancer cachexia. Anal. Biochem. 2020, 606, 113877. [Google Scholar] [CrossRef]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Pro-inflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Sirniö, P.; Väyrynen, J.P.; Klintrup, K.; Mäkelä, J.; Karhu, T.; Herzig, K.H.; Minkkinen, I.; Mäkinen, M.J.; Karttunen, T.J.; Tuomisto, A. Alterations in serum amino-acid profile in the progression of colorectal cancer: Associations with systemic inflammation, tumour stage and patient survival. Br. J. Cancer 2019, 120, 238–246. [Google Scholar] [CrossRef]
- Ohmori, H.; Kawahara, I.; Mori, T.; Nukaga, S.; Luo, Y.; Kishi, S.; Fujiwara-Tani, R.; Mori, S.; Goto, K.; Sasaki, T.; et al. Evaluation of Parameters for Cancer-Induced Sarcopenia in Patients Autopsied after Death from Colorectal Cancer. Pathobiology 2019, 86, 306–314. [Google Scholar] [CrossRef]
- Richards, C.H.; Roxburgh, C.S.; MacMillan, M.T.; Isswiasi, S.; Robertson, E.G.; Guthrie, G.K.; Horgan, P.G.; McMillan, D.C. The relationships between body composition and the systemic inflammatory response in patients with primary operable colorectal cancer. PLoS ONE 2012, 7, e41883. [Google Scholar] [CrossRef] [PubMed]
- Malietzis, G.; Johns, N.; Al-Hassi, H.O.; Knight, S.C.; Kennedy, R.H.; Fearon, K.C.; Aziz, O.; Jenkins, J.T. Low Muscularity and Myosteatosis Is Related to the Host Systemic Inflammatory Response in Patients Undergoing Surgery for Colorectal Cancer. Ann. Surg. 2016, 263, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Abe Vicente, M.; Donizetti Silva, T.; Barão, K.; Vitor Felipe, A.; Oyama Missae, L.; Manoukian Forones, N. The influence of nutritional status and disease on adiponectin and TNF-α; levels in colorectal cancer patients. Nutr. Hosp. 2014, 30, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Nishijima, Y.; Asp, M.L.; Stout, M.B.; Reiser, P.J.; Belury, M.A. Cardiac alterations in cancer-induced cachexia in mice. Int. J. Oncol. 2010, 37, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Strassmann, G.; Jacob, C.O.; Evans, R.; Beall, D.; Fong, M. Mechanisms of experimental cancer cachexia. Interaction between mononuclear phagocytes and colon-26 carcinoma and its relevance to IL-6-mediated cancer cachexia. J. Immunol. 1992, 148, 3674–3678. [Google Scholar]
- Tsoli, M.; Schweiger, M.; Vanniasinghe, A.S.; Painter, A.; Zechner, R.; Clarke, S.; Robertson, G. Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PLoS ONE 2014, 9, e92966. [Google Scholar] [CrossRef]
- Arora, G.; Gupta, A.; Guo, T.; Gandhi, A.; Laine, A.; Williams, D.; Ahn, C.; Iyengar, P.; Infante, R. JAK Inhibitors Suppress Cancer Cachexia-Associated Anorexia and Adipose Wasting in Mice. JCSM Rapid Commun. 2020, 3, 115–128. [Google Scholar] [CrossRef]
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E995–E1010. [Google Scholar] [CrossRef] [PubMed]
- Celik, A.; Kano, Y.; Tsujinaka, S.; Okada, S.; Takao, K.; Takagi, M.; Chohnan, S.; Soda, K.; Kawakami, M.; Konishi, F. Decrease in malonyl-CoA and its background metabolic alterations in murine model of cancer cachexia. Oncol. Rep. 2009, 21, 1105–1111. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, M.; Chen, J.; Wang, L.; Peng, X.; Zhang, X.; Wang, H.; Wang, A.; Zhao, D.; Pang, D.; et al. Abnormality of hepatic triglyceride metabolism in ApcMin/+ mice with colon cancer cachexia. Life Sci. 2019, 227, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.H.; Flegg, K.M.; Roxburgh, C.S.; Going, J.J.; Mohammed, Z.; Horgan, P.G.; McMillan, D.C. The relationships between cellular components of the peritumoural inflammatory response, clinicopathological characteristics and survival in patients with primary operable colorectal cancer. Br. J. Cancer 2012, 106, 2010–2015. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-Infiltrating Immune Cells Act as a Marker for Prognosis in Colorectal Cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef] [PubMed]
- Biasi, F.; Guina, T.; Maina, M.; Nano, M.; Falcone, A.; Aroasio, E.; Saracco, G.M.; Papotti, M.; Leonarduzzi, G.; Poli, G. Progressive increase of matrix metalloprotease-9 and interleukin-8 serum levels during carcinogenic process in human colorectal tract. PLoS ONE 2012, 7, e41839. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell. Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef]
- Ong, S.M.; Tan, Y.C.; Beretta, O.; Jiang, D.; Yeap, W.H.; Tai, J.J.; Wong, W.C.; Yang, H.; Schwarz, H.; Lim, K.H.; et al. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur. J. Immunol. 2012, 42, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Waniczek, D.; Lorenc, Z.; Śnietura, M.; Wesecki, M.; Kopec, A.; Muc-Wierzgoń, M. Tumor-Associated Macrophages and Regulatory T Cells Infiltration and the Clinical Outcome in Colorectal Cancer. Arch. Immunol. Ther. Exp. 2017, 65, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yao, S.; Hu, Y.; Feng, Y.; Li, M.; Bian, Z.; Zhang, J.; Qin, Y.; Qi, X.; Zhou, L.; et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Sun, Y.M. The IL-6/JAK/STAT3 pathway: Potential therapeutic strategies in treating colorectal cancer (Review). Int. J. Oncol. 2014, 44, 1032–1040. [Google Scholar] [CrossRef]
- Strassmann, G.; Masui, Y.; Chizzonite, R.; Fong, M. Mechanisms of experimental cancer cachexia. Local involvement of IL-1 in colon-26 tumor. J. Immunol. 1993, 150, 2341–2345. [Google Scholar]
- Mohebbi, B.; Ashtibaghaei, K.; Hashemi, M.; Hashemi, M.; Asadzadeh Aghdaei, H.; Zali, M.R. Conditioned Medium from Cultured Colorectal Cancer Cells Affects Peripheral Blood Mononuclear Cells Inflammatory Phenotype in Vitro. Iran. J. Med. Sci. 2019, 44, 334–341. [Google Scholar] [CrossRef]
- Inaba, S.; Hinohara, A.; Tachibana, M.; Tsujikawa, K.; Fukada, S.I. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS ONE 2018, 13, e0205467. [Google Scholar] [CrossRef]
- Duong, L.; Radley-Crabb, H.G.; Gardner, J.K.; Tomay, F.; Dye, D.E.; Grounds, M.D.; Pixley, F.J.; Nelson, D.J.; Jackaman, C. Macrophage Depletion in Elderly Mice Improves Response to Tumor Immunotherapy, Increases Anti-tumor T Cell Activity and Reduces Treatment-Induced Cachexia. Front. Genet. 2018, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Reissfelder, C.; Stamova, S.; Gossmann, C.; Braun, M.; Bonertz, A.; Walliczek, U.; Grimm, M.; Rahbari, N.N.; Koch, M.; Saadati, M.; et al. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. J. Clin. Investig. 2015, 125, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Angelova, M.; Charoentong, P.; Hackl, H.; Trajanoski, Z. The colorectal cancer immune paradox revisited. Oncoimmunology 2015, 5, e1078058. [Google Scholar] [CrossRef][Green Version]
- Richards, C.H.; Roxburgh, C.S.; Powell, A.G.; Foulis, A.K.; Horgan, P.G.; McMillan, D.C. The clinical utility of the local inflammatory response in colorectal cancer. Eur. J. Cancer 2014, 50, 309–319. [Google Scholar] [CrossRef]
- Miller, S.; Senior, P.V.; Prakash, M.; Apostolopoulos, V.; Sakkal, S.; Nurgali, K. Leukocyte populations and IL-6 in the tumor microenvironment of an orthotopic colorectal cancer model. Acta Biochim. Biophys. Sin. 2016, 48, 334–341. [Google Scholar] [CrossRef]
- Li, J.; Xu, J.; Yan, X.; Jin, K.; Li, W.; Zhang, R. Targeting Interleukin-6 (IL-6) Sensitizes Anti-PD-L1 Treatment in a Colorectal Cancer Preclinical Model. Med. Sci. Monit. 2018, 24, 5501–5508. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Nezu, T.; Kanou, H.; Abe, H.; Takekawa, M.; Fukuzawa, M. Decreased production of interleukin-12 and type 2 immune responses are marked in cachectic patients with colorectal and gastric cancer. J. Clin. Gastroenterol. 2002, 34, 416–420. [Google Scholar] [CrossRef]
- Tanner, S.M.; Daft, J.G.; Hill, S.A.; Martin, C.A.; Lorenz, R.G. Altered T-Cell Balance in Lymphoid Organs of a Mouse Model of Colorectal Cancer. J. Histochem. Cytochem. 2016, 64, 753–767. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Cancer-related circulating and tumor-associated neutrophils—Subtypes, sources and function. FEBS J. 2018, 285, 4316–4342. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Aoki, T.; Tsuruyama, T.; Narumiya, S. Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015, 75, 2822–2832. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.L.; Chen, J.W.; Li, M.; Xiao, Y.B.; Fu, J.; Zeng, Y.X.; Cai, M.Y.; Xie, D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS ONE 2012, 7, e30806. [Google Scholar] [CrossRef]
- Governa, V.; Trella, E.; Mele, V.; Tornillo, L.; Amicarella, F.; Cremonesi, E.; Muraro, M.G.; Xu, H.; Droeser, R.; Däster, S.R.; et al. The Interplay Between Neutrophils and CD8+ T Cells Improves Survival in Human Colorectal Cancer. Clin. Cancer Res. 2017, 23, 3847–3858. [Google Scholar] [CrossRef]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, M.; Soda, K.; Gin, P.T.; Kai, T.; Konishi, F.; Kawakami, M. Erythropoietin attenuates cachectic events and decreases production of interleukin-6, a cachexia-inducing cytokine. Cytokine 2005, 32, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Winfield, R.D.; Delano, M.J.; Pande, K.; Scumpia, P.O.; Laface, D.; Moldawer, L.L. Myeloid-derived suppressor cells in cancer cachexia syndrome: A new explanation for an old problem. JPEN J. Parenter. Enteral. Nutr. 2008, 32, 651–655. [Google Scholar] [CrossRef]
- Chouaib, S.; Umansky, V.; Kieda, C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Contemp. Oncol. 2018, 22, 7–13. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv. Sci. 2019, 6, 1901278. [Google Scholar] [CrossRef] [PubMed]
- Ohki, S.; Shibata, M.; Gonda, K.; Machida, T.; Shimura, T.; Nakamura, I.; Ohtake, T.; Koyama, Y.; Suzuki, S.; Ohto, H.; et al. Circulating myeloid-derived suppressor cells are increased and correlate to immune suppression, inflammation and hypoproteinemia in patients with cancer. Oncol. Rep. 2012, 28, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gonda, K.; Shibata, M.; Shimura, T.; Machida, T.; Suzuki, S.; Nakamura, I.; Ohki, S.; Sakurai, K.; Ohto, H.; Tomita, R.; et al. Serum Soluble Interleukin-2 Receptor is Increased in Malnourished and Immunosuppressed Patients with Gastric and Colorectal Cancer: Possible Influence of Myeloid-Derived Suppressor Cells. World J. Oncol. 2012, 3, 158–164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakamura, I.; Shibata, M.; Gonda, K.; Yazawa, T.; Shimura, T.; Anazawa, T.; Suzuki, S.; Sakurai, K.; Koyama, Y.; Ohto, H.; et al. Serum levels of vascular endothelial growth factor are increased and correlate with malnutrition, immunosuppression involving MDSCs and systemic inflammation in patients with cancer of the digestive system. Oncol. Lett. 2013, 5, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Batista, M.L., Jr.; Olivan, M.; Alcantara, P.S.; Sandoval, R.; Peres, S.B.; Neves, R.X.; Silverio, R.; Maximiano, L.F.; Otoch, J.P.; Seelaender, M. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine 2013, 61, 532–539. [Google Scholar] [CrossRef]
- Neto, N.I.P.; Murari, A.S.P.; Oyama, L.M.; Otoch, J.P.; Alcântara, P.S.M.; Tokeshi, F.; Figuerêdo, R.G.; Alves, M.J.; Lima, J.D.C.C.; Matos-Neto, E.M.; et al. Peritumoural adipose tissue pro-inflammatory cytokines are associated with tumoural growth factors in cancer cachexia patients. J. Cachexia Sarcopenia Muscle 2018, 9, 1101–1108. [Google Scholar] [CrossRef]
- De Matos-Neto, E.M.; Lima, J.D.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.; Radloff, K.; das Neves, R.X.; Camargo, R.G.; Maximiano, L.F.; Tokeshi, F.; et al. Systemic Inflammation in Cachexia—Is Tumor Cytokine Expression Profile the Culprit? Front. Immunol. 2015, 6, 629. [Google Scholar] [CrossRef]
- Chang, M.L.; Yang, Z.; Yang, S.S. Roles of Adipokines in Digestive Diseases: Markers of Inflammation, Metabolic Alteration and Disease Progression. Int. J. Mol. Sci. 2020, 21, 8308. [Google Scholar] [CrossRef]
- Silvério, R.; Lira, F.S.; Oyama, L.M.; Oller do Nascimento, C.M.; Otoch, J.P.; Alcântara, P.S.M.; Batista, M.L., Jr.; Seelaender, M. Lipases and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017, 16, 159. [Google Scholar] [CrossRef]
- Batista, M.L., Jr.; Henriques, F.S.; Neves, R.X.; Olivan, M.R.; Matos-Neto, E.M.; Alcântara, P.S.; Maximiano, L.F.; Otoch, J.P.; Alves, M.J.; Seelaender, M. Cachexia-associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 2016, 7, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.; Yehuda-Shnaidman, E. Putative role of adipose tissue in growth and metabolism of colon cancer cells. Front. Oncol. 2014, 4, 164. [Google Scholar] [CrossRef] [PubMed]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity-Cancer Link-A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Tseng, C.; Zhang, Y.; Sirin, O.; Corn, P.G.; Li-Ning-Tapia, E.M.; Troncoso, P.; Davis, J.; Pettaway, C.; Ward, J.; et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 2016, 7, 11674. [Google Scholar] [CrossRef] [PubMed]
- Welte, G.; Alt, E.; Devarajan, E.; Krishnappa, S.; Jotzu, C.; Song, Y.H. Interleukin-8 derived from local tissue-resident stromal cells promotes tumor cell invasion. Mol. Carcinog. 2012, 51, 861–868. [Google Scholar] [CrossRef]
- Fenton, J.I.; Hursting, S.D.; Perkins, S.N.; Hord, N.G. Interleukin-6 production induced by leptin treatment promotes cell proliferation in an Apc (Min/+) colon epithelial cell line. Carcinogenesis 2006, 27, 1507–1515. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Denton, A.E.; Roberts, E.W.; Fearon, D.T. Stromal Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2018, 1060, 99–114. [Google Scholar] [CrossRef]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Bartolomé, R.A.; Mendes, M.; Barderas, R.; Fernandez-Aceñero, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; de Herreros, A.G.; et al. Proteome profiling of cancer-associated fibroblasts identifies novel pro-inflammatory signatures and prognostic markers for colorectal cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, D.; Tian, J.; Gao, A.; Shen, Y.; Ren, X.; Li, X.; Jiang, G.; Dong, T. Tumor necrosis factor receptor 2/AKT and ERK signaling pathways contribute to the switch from fibroblasts to CAFs by progranulin in microenvironment of colorectal cancer. Oncotarget 2017, 8, 26323–26333. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Shang, Y.; Sun, F.; Dong, X.; Niu, J.; Li, F. Interleukin-6 Promotes Epithelial-Mesenchymal Transition and Cell Invasion through Integrin β6 Upregulation in Colorectal Cancer. Oxid. Med. Cell Longev. 2020, 2020, 8032187. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Ao, T.; Sugiura, T.; Yonemura, K.; Shiraishi, T.; Kajiwara, Y.; Okamoto, K.; Shinto, E.; Okada, Y.; Ueno, H. Expression and Function of a Disintegrin and Metalloproteinases in Cancer-Associated Fibroblasts of Colorectal Cancer. Digestion 2020, 101, 18–24. [Google Scholar] [CrossRef]
- Li, T.; Yi, S.; Liu, W.; Jia, C.; Wang, G.; Hua, X.; Tai, Y.; Zhang, Q.; Chen, G. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med. Oncol. 2013, 30, 663. [Google Scholar] [CrossRef]
- Cabal-Manzano, R.; Bhargava, P.; Torres-Duarte, A.; Marshall, J.; Bhargava, P.; Wainer, I.W. Proteolysis-inducing factor is expressed in tumours of patients with gastrointestinal cancers and correlates with weight loss. Br. J. Cancer 2001, 84, 1599–1601. [Google Scholar] [CrossRef]
- Todorov, P.; Cariuk, P.; McDevitt, T.; Coles, B.; Fearon, K.; Tisdale, M. Characterization of a cancer cachectic factor. Nature 1996, 379, 739–742. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Fearon, K.C. Cachexia, survival and the acute phase response. Curr. Opin. Support Palliat. Care 2008, 2, 267–274. [Google Scholar] [CrossRef]
- Cui, G.; Yuan, A.; Sun, Z.; Zheng, W.; Pang, Z. IL-1β/IL-6 network in the tumor microenvironment of human colorectal cancer. Pathol. Res. Pract. 2018, 214, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, K.; Mukaida, N.; Harada, A.; Kuno, K.; Akiyama, M.; Nakashima, E.; Fujioka, N.; Mai, M.; Kasahara, T.; Fujimoto-Ouchi, K.; et al. Molecular analysis of the cytokine network involved in cachexia in colon 26 adenocarcinoma-bearing mice. Cancer Res. 1995, 55, 921–927. [Google Scholar] [PubMed]
- Toyoshima, Y.; Kitamura, H.; Xiang, H.; Ohno, Y.; Homma, S.; Kawamura, H.; Takahashi, N.; Kamiyama, T.; Tanino, M.; Taketomi, A. IL6 Modulates the Immune Status of the Tumor Microenvironment to Facilitate Metastatic Colonization of Colorectal Cancer Cells. Cancer Immunol. Res. 2019, 7, 1944–1957. [Google Scholar] [CrossRef]
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal Cell PD-L1 Inhibits CD8+ T-cell Antitumor Immune Responses and Promotes Colon Cancer. Cancer Immunol. Res. 2018, 6, 1426–1441. [Google Scholar] [CrossRef]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.; et al. Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef]
- Schäfer, M.; Oeing, C.U.; Rohm, M.; Baysal-Temel, E.; Lehmann, L.H.; Bauer, R.; Volz, H.C.; Boutros, M.; Sohn, D.; Sticht, C.; et al. Ataxin-10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol. Metab. 2015, 5, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Seto, D.N.; Kandarian, S.C.; Jackman, R.W. A Key Role for Leukemia Inhibitory Factor in C26 Cancer Cachexia. J. Biol. Chem. 2015, 290, 19976–19986. [Google Scholar] [CrossRef]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed]
- Yakovenko, A.; Cameron, M.; Trevino, J.G. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 2018, 10, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.E.; Dasgupta, A.; King, R.J.; Abrego, J.; Attri, K.S.; Murthy, D.; Shukla, S.K.; Singh, P.K. JNK signaling contributes to skeletal muscle wasting and protein turnover in pancreatic cancer cachexia. Cancer Lett. 2020, 491, 70–77. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Jiang, Z.W.; Tian, J.; Jiang, J.; Li, N.; Li, J.S. Role of NF-kappaB and cytokine in experimental cancer cachexia. World J. Gastroenterol. 2003, 9, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, Q.; Zhang, H.; Wan, L.; Xin, B.; Cao, Y.; Zhang, J.; Guo, C. Cryptotanshinone prevents muscle wasting in CT26-induced cancer cachexia through inhibiting STAT3 signaling pathway. J. Ethnopharmacol. 2020, 260, 113066. [Google Scholar] [CrossRef]
- Rydén, M.; Arner, P. Tumour necrosis factor-alpha in human adipose tissue—From signalling mechanisms to clinical implications. J. Intern. Med. 2007, 262, 431–438. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef]
- Manne, N.D.; Lima, M.; Enos, R.T.; Wehner, P.; Carson, J.A.; Blough, E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol. 2013, 42, 2134–2140. [Google Scholar] [CrossRef] [PubMed]
- De Castro, G.S.; Simoes, E.; Lima, J.D.C.C.; Ortiz-Silva, M.; Festuccia, W.T.; Tokeshi, F.; Alcântara, P.S.; Otoch, J.P.; Coletti, D.; Seelaender, M. Human Cachexia Induces Changes in Mitochondria, Autophagy and Apoptosis in the Skeletal Muscle. Cancers 2019, 11, 1264. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Camuzard, O.; Santucci-Darmanin, S.; Carle, G.F.; Pierrefite-Carle, V. Autophagy in the crosstalk between tumor and microenvironment. Cancer Lett. 2020, 490, 143–153. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Pavlides, S.; Chiavarina, B.; Bonuccelli, G.; Casey, T.; Tsirigos, A.; Migneco, G.; Witkiewicz, A.; Balliet, R.; et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010, 9, 4297–4306. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; López-Soriano, F.J. Anti-inflammatory therapies in cancer cachexia. Eur. J. Pharmacol. 2011, 668 (Suppl. 1), S81–S86. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Counteracting inflammation: A promising therapy in cachexia. Crit. Rev. Oncog. 2012, 17, 253–262. [Google Scholar] [CrossRef]
- Ebner, N.; Werner, C.G.; Doehner, W.; Anker, S.D.; von Haehling, S. Recent developments in the treatment of cachexia: Highlights from the 6th Cachexia Conference. J. Cachexia Sarcopenia Muscle 2012, 3, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Prado, B.L.; Qian, Y. Anti-cytokines in the treatment of cancer cachexia. Ann. Palliat. Med. 2019, 8, 67–79. [Google Scholar] [CrossRef]
- Chen, S.Z.; Qiu, Z.G. Combined treatment with GH, insulin, and indomethacin alleviates cancer cachexia in a mouse model. J. Endocrinol. 2011, 208, 131–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molfino, A.; Amabile, M.I.; Rossi Fanelli, F.; Muscaritoli, M. Novel therapeutic options for cachexia and sarcopenia. Expert. Opin. Biol. Ther. 2016, 16, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Hickish, T.; Andre, T.; Wyrwicz, L.; Saunders, M.; Sarosiek, T.; Kocsis, J.; Nemecek, R.; Rogowski, W.; Lesniewski-Kmak, K.; Petruzelka, L.; et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017, 18, 192–201. [Google Scholar] [CrossRef]
- O’Sullivan Coyne, G.; Burotto, M. MABp1 for the treatment of colorectal cancer. Expert Opin. Biol. Ther. 2017, 17, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.S.; Li, X.; Cao, Y.Z.; Zhang, C.C.; Yang, F.; Shi, Y.M.; Peng, L.M. Clinical studies on the treatment of cancer cachexia with megestrol acetate plus thalidomide. Chemotherapy 2012, 58, 461–467. [Google Scholar] [CrossRef]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. Control of food intake and muscle wasting in cachexia. Int. J. Biochem. Cell Biol. 2013, 45, 2179–2185. [Google Scholar] [CrossRef]
- De Waele, E.; Demol, J.; Caccialanza, R.; Cotogni, P.; Spapen, H.; Malbrain, M.L.; De Grève, J.; Pen, J.J. Unidentified cachexia patients in the oncologic setting: Cachexia UFOs do exist. Nutrition 2019, 63–64, 200–204. [Google Scholar] [CrossRef]
- Blackwood, H.A.; Hall, C.C.; Balstad, T.R.; Solheim, T.S.; Fallon, M.; Haraldsdottir, E.; Laird, B.J. A systematic review examining nutrition support interventions in patients with incurable cancer. Support Care Cancer 2020, 28, 1877–1889. [Google Scholar] [CrossRef]
- Van de Worp, W.R.P.H.; Schols, A.M.W.J.; Theys, J.; van Helvoort, A.; Langen, R.C.J. Nutritional Interventions in Cancer Cachexia: Evidence and Perspectives from Experimental Models. Front. Nutr. 2020, 7, 601329. [Google Scholar] [CrossRef] [PubMed]
- Hamauchi, S.; Furuse, J.; Takano, T.; Munemoto, Y.; Furuya, K.; Baba, H.; Takeuchi, M.; Choda, Y.; Higashiguchi, T.; Naito, T.; et al. A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia. Cancer 2019, 125, 4294–4302. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Arai, H.; Inui, A. The regulatory approval of anamorelin for treatment of cachexia in patients with non-small cell lung cancer, gastric cancer, pancreatic cancer, and colorectal cancer in Japan: Facts and numbers. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Summermatter, S.; Jourdain, M.; Melly, S.; Minetti, G.C.; Lach-Trifilieff, E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet. Muscle 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Tahaghoghi-Hajghorbani, S.; Ebrahimzadeh, M.A.; Rafiei, A.; Golpour, M.; Hosseini-Khah, Z.; Akhtari, J. Improvement of chemotherapy through reducing of cachexia by using Citrus unshiu peel extract. J. Ethnopharmacol. 2019, 242, 111929. [Google Scholar] [CrossRef] [PubMed]
- Shadfar, S.; Couch, M.E.; McKinney, K.A.; Weinstein, L.J.; Yin, X.; Rodríguez, J.E.; Guttridge, D.C.; Willis, M. Oral resveratrol therapy inhibits cancer-induced skeletal muscle and cardiac atrophy in vivo. Nutr. Cancer 2011, 63, 749–762. [Google Scholar] [CrossRef]


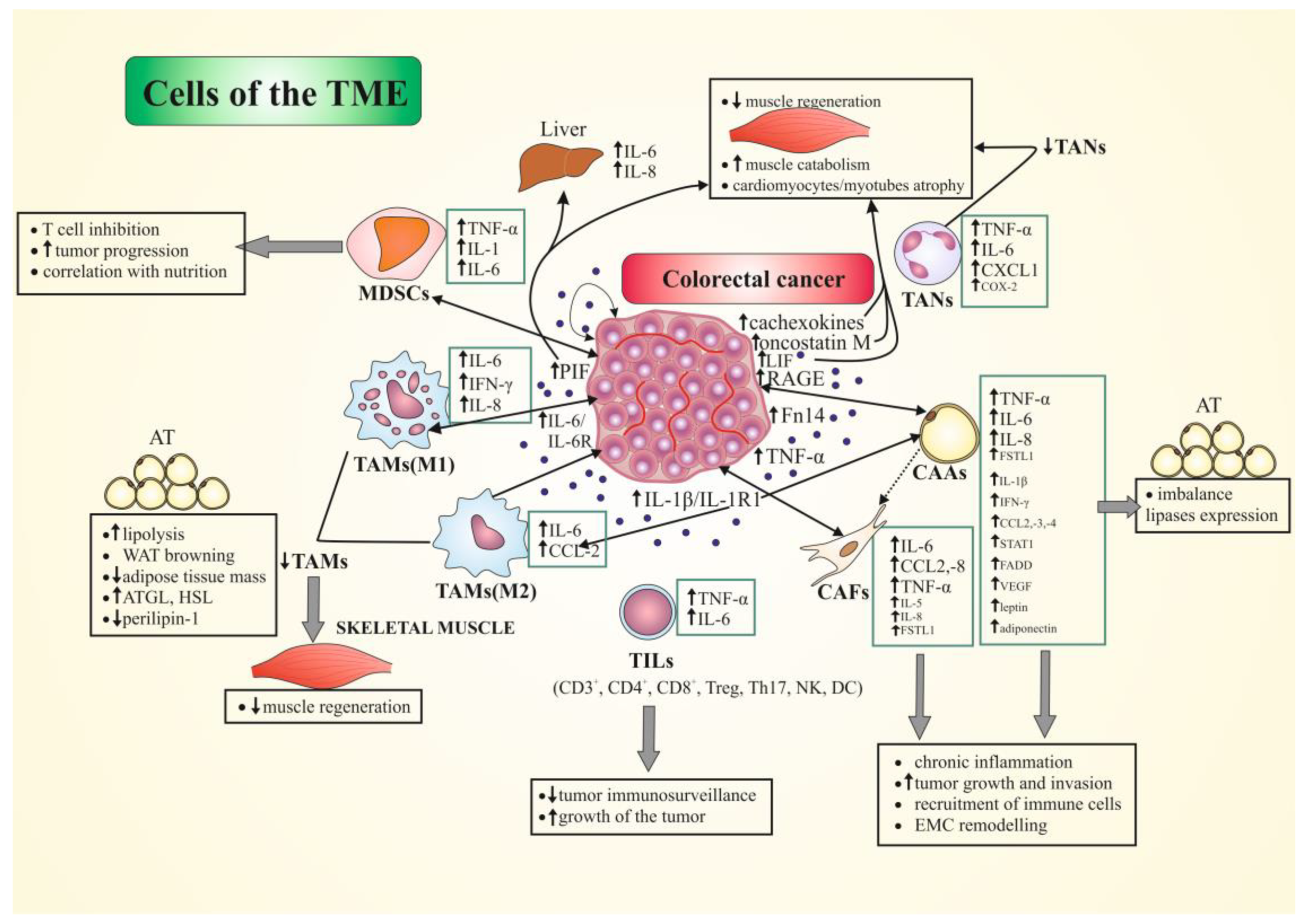{kind=link}
| Procachectic Mediator | Model of the Study | Target Tissue | Mechanism of Action | Ref. |
|---|---|---|---|---|
| TNF-α | Murine C2C12 and primary myoblasts; mouse colon-26 cells (C26); various in vitro and in vivo mouse models of CC; patients with CC | Muscle | (i) catabolic effects; (ii) TNF-α + IFN-γ led to ↓expression of myosin via RNA-dependent mechanism in myotubes and muscle tissue; (iii) UPS pathway contribution | [44] |
| Adipose; muscle | (i) catabolic effects; (ii) loss of AT and proteolysis, while causing ↓protein, lipid, and glycogen synthesis; (iii) ↓lipoprotein lipase; ↑gluconeogenesis; (iv) ↓protein synthesis via ↓in the active eIF4F complex; (v) serum levels do not correlate with weight loss | [5,41] | ||
| CRC patients with different stages of CC; BALB/c male mice; C26/clone 20 cells | Adipose | was positively linked to FFA in early- but not late-stage CC | [60] | |
| cancer patients with CC; WSC; C | ↑level in CC patients vs. C | [61] | ||
| CRC tissue samples | Muscle | (i) negative correlation with SMI and SDS-MYL1; (ii) positive correlation with HMGB1 | [67] * | |
| primary operable CRC and control | (i) ↑level was correlated with CRC staging; (ii) patients with the greatest nutritional deficit exhibited ↑levels of adipocytokines | [70] * | ||
| PIF | various mouse models of CC; cancer patients with CC | Muscle | (i) catabolic effect on skeletal muscle; (ii) ↓protein synthesis via ↑phosphorylation of the eIF2 on the alpha-subunit; (iii) its presence is indicative of weight loss; (iv) its presence in the urine of CC patients is indicative of weight loss | [41] |
| C2C12 mouse myoblasts; MAC16 tumors | Muscle | ↑Ca+2i, initiating a signaling cascade that leads to a ↓protein synthesis and ↑ in protein degradation | [45] | |
| IL-1 (α and β) | C2C12 myoblasts, mature C2C12 myotubes model | Muscle | (i) an oxidant and Akt/FOXO-independent mechanism to activate p38 MAPK, ↑NF-κB signaling; (ii) ↑expression of Atrogin 1/MAFbx and MuRF1, and ↓myofibrillar protein in differentiated myotubes; (iii) the direct mechanisms in human CC tissue wasting are not recognized | [50] |
| IFN-γ | Various in vitro and in vivo mouse models of CC | Adipose; muscle | (i) with TNF-α led to ↓expression of myosin via RNA-dependent mechanism in myotubes and muscle tissue; (ii) UPS pathway contribution; (iii) changes in expression and signaling may be perceived at stages preceding refractory cachexia | [44,51] |
| cancer patients with CC; WSC; C | positive correlation with plasma FA profile | [61] | ||
| IL-6 | ApcMin/+ mice; C2C12 cells | Muscle | (i) ↑muscle wasting; (ii) altered the expression of proteins regulating mitochondrial biogenesis and fusion; (iii) ↓in mitochondrial content during the progression of cachexia; (iv) directly ↑FIS1 expression in muscle cells; (v) ↑indices of ROS in myotubes | [56] |
| mature C2C12 myotubes model; in vitro and in vivo mouse model of CC | Muscle | STAT3 activation is a common feature of muscle wasting, muscle fiber atrophy and exacerbated wasting in CC | [53] | |
| Mouse model of CC; C26 cells; human cancerous-tissue samples | Adipose | ↑expression of UPC1 in WAT, which affects the mitochondrial respiration, uncoupling it toward thermogenesis instead of ATP synthesis, which results in ↑lipid mobilization and energy spending in CC | [59] | |
| CRC patients with different stages of CC; BALB/c male mice; C26/clone 20 cells | Adipose | (i) regulating WAT lipolysis in early-stage cachexia and browning in late-stage cachexia; (ii) correlation with serum FFA | [60] | |
| cancer patients with CC; WSC; C | ↑level in CC patients vs. control | [61] | ||
| C26 model of CC | Adipose | (i) ↓AT mass, ↑AT lipolysis, and a 5-fold ↑ in FFA plasma levels; (ii) activation of IL-6 signaling in WAT through a 3-fold ↑ in pSTAT3 and high SOCS3 gene expression levels | [73] * | |
| CD2F1 mice inoculated with C26 cells and vehicle | Muscle | (i) ↑IL-6, IL-6R, and F4/80 in the heart of tumor-bearing vs. control; (ii) ↑fibrosis, disrupted myocardial structure, and altered composition of contractile proteins | [71] * | |
| IL-8 | cancer patients with CC; WSC; C | (i) ↑level in CC patients vs. control and in WSC vs. control; (ii) positive correlation with plasma FA profile | [61] |
| TME Cell Type | Secreted Mediators | Intercellular Cooperation | Signaling Pathways | Potential Role in Cachexia | Ref. |
|---|---|---|---|---|---|
| TAMs | M1 cells: ↑IL-6, IFN-γ, CXCL8/IL-8, CCL2/MCP1; M2 cells: ↑IL-6, CCL2; Monocytes: ↑IL-6, IL-12b, IFN-γ; IL-1; ↓IL-4, IL-10, TNF-α | Other immune cells (e.g., T cells); CRC cells (including colon adenocarcinoma C26 cells) | IL-1/IL-1R1; IL-6/STAT3; PGE2/EP;JAK2/STAT3/miR-506-3p/FOXQ1; IL-6R/STAT3/ miR-204-5p | (i) M1: pro-inflammatory in vitro and in vivo, promoting of type-1 T-cell response, with a tumor-suppressive role, and inhibiting of proliferation of tumor cells; (ii) ↑cellular interaction between IL-1R-expressing tumor cells and TAMs; (iii) M2: regulate of EMT, ↑migration and invasion of CRC cells, induce of chemoresistance of CRC cells; (iv) ↓TAMs number correlated with disturbed skeletal-muscle regenerative ability in CC through ↓CCL2-5, CXCL3 levels | [23,53,72,83,85,86,87,88,89,90] |
| TILs | T cells: ↑TNF-α, IL-6; PBMCs: ↑IL-6, IL-4, IL-10; ↓IL-12 | Other immune cells (e.g., macrophages); CRC cells | (i) ↑tumor growth; (ii) ↑↑IL-6 correlated with ↓CD8+ and CD4+ lymphocytes, and ↑MDSCs and Treg/suppressor T cells in tumor; (iii) correlation between ↑↑IL-6 and ↑↑PD-L1; (iv) ↓intestinal-tumor immunosurveillance | [92,96,97,98] | |
| TANs | ↑TNF-α, IL-6, CXCL1, COX-2 | Other immune cells (e.g., T cells); CRC cells | TGF-β; IFN-γ; PGE2/EP2 | (i) EP2 stimulate TANs to ↑expression of cytokines/chemokines and other pro-inflammatory genes; (ii) role in angiogenesis, cell migration, invasion, and metastasis; (iii) ↑CD8+ T-cell activation, proliferation, and cytokine release; (iv) ↑CD45RO/CD62L T-cell number; (v) ↓TANs number correlated with disturbed muscle regenerative ability in CC through ↓CCL2-5, CXCL3 levels | [20,23,90,100,101,102,104] |
| MDSCs | ↑TNF-α, IL-1, IL-6 | Other immune cells (e.g., T cells); CRC cells | (i) ↓T cell functions after HIF-1α stimulation; (ii) promote CRC cell stemness and progression via exosomes enriched in S100A9; (iii) suppress cell-mediated immune response; (iv) circulating cells correlate with sIL-2R levels and nutritional parameters | [108,110,111,113] | |
| CAAs | pAT: ↑TNF-α, STAT1, FADD; sAT: ↑TNF-α, IL-1β; CCL2/MCP-1 gene expression; ↑CCL3/MIP-1α, CCL4, IL-1β and TNF-β protein expression; ↑ATGL and HSL, ↓perilipin AT and plasma: ↑IL-8, TGF-β1, -β2, and β3; IFN-γ, EGF; GM-CSF, HIF-1α; ↑leptin, adiponectin, IGF1, TNF-α, IL-6, IL-8, IL-10, CCL2, VEGF | CRC cells; Cancer myofibroblasts; Immune cells (e.g., macrophages, lymphocytes) | TGF-β; PI3K/JAK/STAT3/ MAPK | (i) recruit of M1 and M2 macrophages; (ii) ↑lipolysis in sAT associated with macrophage infiltration (prevalent reparative inflammatory response), imbalance lipases expression; (iii) ↑of fibrosis of sAT, tumor remodeling, transdifferentiation of fibroblast to myofibroblasts surrounding adipocytes with ↑ECM (iv) imbalance inflammatory cytokine profile; (v) activate cell-cycle regulators; (vi) are reprogrammed into CAFs by cancer cells; (vii) ↑tumor cell growth and invasion | [18,29,30,117,118,120,121,122,123,125,126] |
| CAFs | ↑CCL2, CCL8; FSTL1; IL-6; TNF-α, IL-5, IL-8; ADAMs; COX-2, Wnt genes; PGE2 | CRC cells; CAAs; Immune cell (e.g., NK cells) | TGF-β/Smad; TNFR2/Akt (ERK); VEGF-A; PGE2/EP | (i) metabolically support tumor growth via the local stromal generation of mitochondrial fuels; (ii) role in tissue remodeling and fibrosis/desmoplakia; (iii) imbalance inflammatory cytokine profile; (iv) ↑migration/invasion of CRC cells; (v) role in angiogenesis (vi) ↑ECM synthesis; (vii)regulate expression of inflammation- and growth-related genes and suppress NK cells function | [23,29,30,127,135,138] |
| CRC cells | ↑PIF; progranulin ↑IL-6, IL-6R, IL-1R1, IL-1β; ↑TNF-α; ↑↑Fn14 (receptor of TWEAK); ↑oncostatin M, LIF↑RAGE; ↑cachexokines (e.g., Ataxin-10) | Immune cells (e.g., monocytes/ macrophages, T cells, MDSCs); liver cells; CAAs; CAFs; ECs | JAK/STAT3; LIF/JAK2/STAT3 | (i) ↑protein degradation; (ii) ↑IL-6 and IL-8 production by liver cells; (iii) ↑strong immunosuppression in TME and metastatic colonization of C26 cells via IL-6; (iv) role in muscle wasting, systemic inflammation, release of tumor-derived procachectic factors through RAGE, S100B and HMGB1; (v) ↑atrophy of cardiomyocytes and alter lipid metabolism; (vi) ↑myotubes atrophy; (vi) role in monocytes differentiation toward inflammatory phenotypes; (vii) role in activation of fibroblast to myofibroblasts; (viii) ↑IL-6 production from fibroblasts resulting in ↑integrin β6; (ix) ↑glycolysis in CAFs and ECs, cause T-cell immunosuppression | [53,89,97,118,132,134,136,139,140,142,143,144,146,147,148,149,151] |
| Name of Targeted Agents | Agent/Phase | Population/Animal Model of the Study | Main Effects in Cachexia | Ref. |
|---|---|---|---|---|
| Thalidomide | a derivative of glutamic acid; phase II | Different advanced human cancer types; (i) megestrol acetate (MA) plus thalidomide; (ii) megestrol | MA + thalidomide improved weight, fatigue, quality of life, grip strength, GPS, ECOG performance status; ↓IL-6, and TNF-α in combination arm | [176] |
| MABp1 | first-in-class true human IgG1k MAb against IL-1α; phase III | humans | (i) antitumor; (ii) more patients in MABp1 group achieved the composite primary outcome; (iii) MABp1 group had lower IL-6, less thrombocytosis, and longer survival | [174] |
| Anamorelin (ONO-7643; ANAM) | ghrelin receptor agonist; approved for use in Japan (2020) | humans | (i) improves anorexia and patients’ nutritional status; (ii) ↑serum IGF1 | [181,182] |
| Promising trials on animal models | ||||
| GH, insulin, indomethacin | C26 model, mouse model | alleviated tumor-free body-weight reduction and CC-induced changes in nutritional markers and cytokines, and prolonged survival time | [172] | |
| Anti-Fn14 | antibody against Fn14 | C26 cells, mice, humans | (i) retain body mass; (ii) retain muscle mass, (iii) retain AT | [146] |
| Anti-RAGE | antibody against RAGE | mouse model | lack of RAGE results in reduced serum levels of cachexia-inducing factors, delayed loss of muscle mass and strength, reduced tumor progression, and increased survival | [147] |
| CDD866 | a neutralizing antibody against ActRII | CT-26 mouse colon cancer-induced cachexia model | administration of CDD866 alone or in combination with cisplatin protected from skeletal-muscle weight loss compared to animals receiving only cisplatin | [183] |
| Resveratrol | natural phytoalexin | C26 cells, mouse model | (i) inhibits NF-κB in cancer cells; (ii) inhibits of NF-κB activity in skeletal and heart muscle; (iii) inhibits skeletal and cardiac muscle atrophy induced by C26 tumors | [185] |
| Citrus unshiu Peel Extract (CUPE) | traditional herbal drug | C26 tumor-bearing BALB/c male mice | (i) ↓weight loss, tumor volume, and serum MDA levels; (ii) the combination therapy (CUPE + Dox) leads to ↓↓serum levels of IL-6, TNF-α, IL-1β and tumor volume vs. untreated tumor-bearing mice and Dox groups | [184] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int. J. Mol. Sci. 2021, 22, 1565. https://doi.org/10.3390/ijms22041565
Kasprzak A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. International Journal of Molecular Sciences. 2021; 22(4):1565. https://doi.org/10.3390/ijms22041565
Chicago/Turabian StyleKasprzak, Aldona. 2021. "The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia" International Journal of Molecular Sciences 22, no. 4: 1565. https://doi.org/10.3390/ijms22041565
APA StyleKasprzak, A. (2021). The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. International Journal of Molecular Sciences, 22(4), 1565. https://doi.org/10.3390/ijms22041565