
DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana

 ,
,
Abstract
1. Introduction
2. Results
2.1. Experiment Design and Whole-Genome Bisulfite Sequencing
2.2. Heat Stress induces DNA Demethylation in Genes
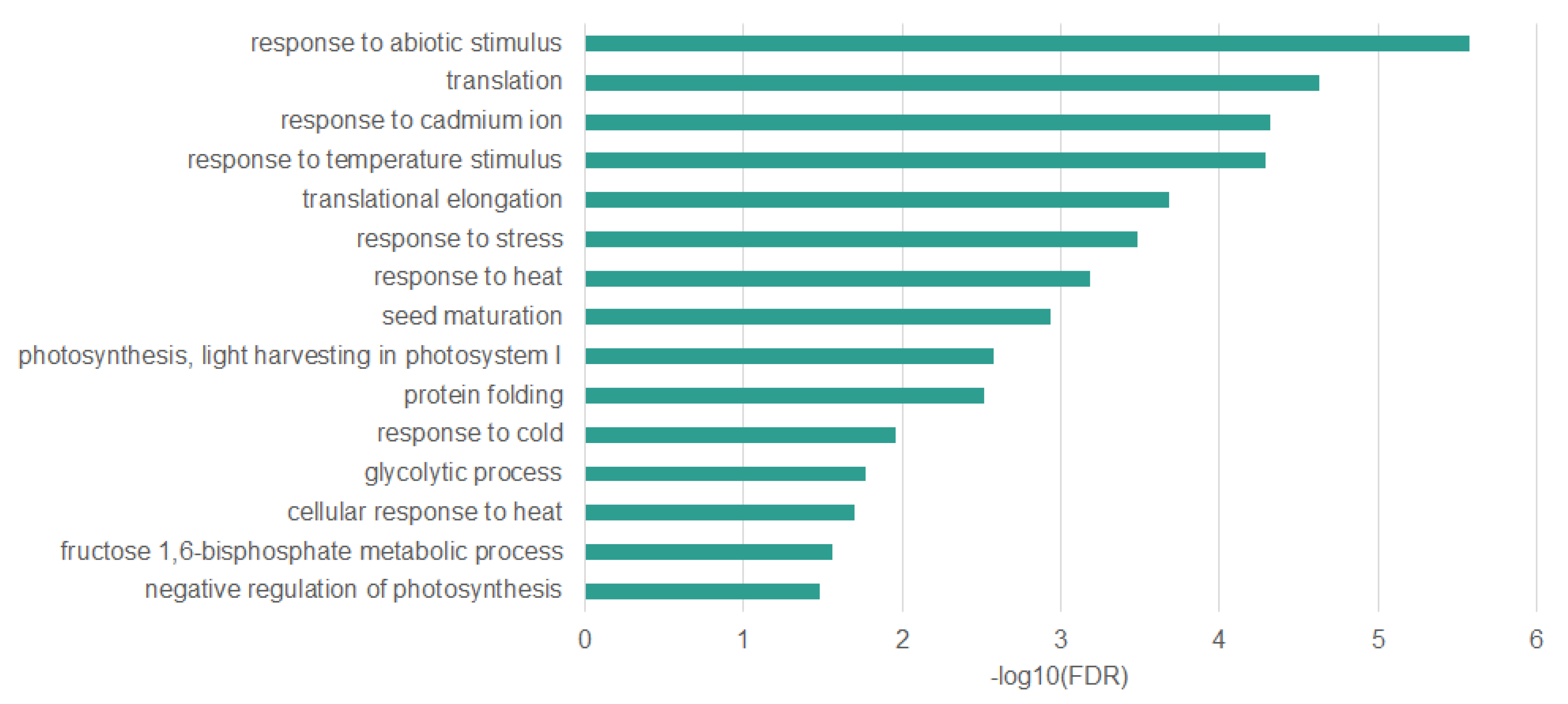
2.3. Genes Undergoing DNA Demethylation under Heat Stress are Associated with the Stress Response
3. Discussion
4. Materials and Methods
4.1. Plant Material, Growth, and Stress Conditions
4.2. Nucleic Acids Isolation
4.3. WGBS Library Preparation and Sequencing
4.4. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMC | differentially methylated cytosine |
| HSP | heat shock protein |
References
- Kumar, A.A.; Mishra, P.; Kumari, K.; Panigrahi, K. Environmental Stress Influencing Plant Development and Flowering. Front Biosci. 2012, 4, 1315–1324. [Google Scholar]
- Ayub, M.; Ashraf, M.Y.; Kausar, A.; Saleem, S.; Anwar, S.; Altay, V.; Ozturk, M. Growth and Physio-Biochemical Responses of Maize (Zea Mays L.) to Drought and Heat Stresses. Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2020, 1–13. [Google Scholar] [CrossRef]
- Nguyen, C.T.T.; Cheong, J.-J.; Nguyen, N.H.; Choi, W.S.; Lee, J.H. Biosynthesis of Essential Oil Compounds in Ocimum Tenuiflorum Is Induced by Abiotic Stresses. Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2020, 1–8. [Google Scholar] [CrossRef]
- Hirayama, T.; Shinozaki, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant J. 2010, 61, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Bita, C.; Gerats, T. Plant tolerance to high temperature in a changing environment: Scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 2013, 4, 273. [Google Scholar] [CrossRef]
- Źróbek-Sokolnik, A. Temperature stress and responses of plants. In Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change; Springer: Cham, Switzerland, 2012; pp. 113–134. [Google Scholar]
- Al-Whaibi, M.H. Plant heat-shock proteins: A mini review. J. King Saud Univ.-Sci. 2011, 23, 139–150. [Google Scholar] [CrossRef]
- Guo, M.; Liu, J.-H.; Ma, X.; Luo, D.-X.; Gong, Z.-H.; Lu, M.-H. The Plant Heat Stress Transcription Factors (HSFs): Structure, Regulation and Function in Response to Abiotic Stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef]
- Goyal, K.; Walton, L.J.; Tunnacliffe, A. LEA proteins prevent protein aggregation due to water stress. Biochem. J. 2005, 388, 151–157. [Google Scholar] [CrossRef]
- Huang, B.; Xu, C. Identification and characterization of proteins associated with plant tolerance to heat stress. J. Integr. Plant Biol. 2008, 50, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Momčilović, I.; Clemente, T.E.; Nersesian, N.; Trick, H.N.; Ristic, Z. Heterologous Expression of a Plastid EF-Tu Reduces Protein Thermal Aggregation and Enhances CO2 Fixation in Wheat (Triticum Aestivum) Following Heat Stress. Plant Mol. Biol. 2008, 68, 277–288. [Google Scholar] [CrossRef]
- Prasad, P.V.; Pisipati, S.; Mutava, R.; Tuinstra, M. Sensitivity of Grain Sorghum to High Temperature Stress during Reproductive Development. Crop Sci. 2008, 48, 1911–1917. [Google Scholar] [CrossRef]
- Pressman, E.; Peet, M.M.; Pharr, D.M. The Effect of Heat Stress on Tomato Pollen Characteristics Is Associated with Changes in Carbohydrate Concentration in the Developing Anthers. Ann. Bot. 2002, 90, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef] [PubMed]
- Mohtat, D.; Susztak, K. Fine tuning gene expression: The epigenome. Semin. Nephrol. 2010, 30, 468–476. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204. [Google Scholar] [CrossRef]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351. [Google Scholar] [CrossRef]
- Rocha, P.S.C.F.; Sheikh, M.; Melchiorre, R.; Fagard, M.; Boutet, S.; Loach, R.; Moffatt, B.; Wagner, C.; Vaucheret, H.; Furner, I. The Arabidopsis HOMOLOGY-DEPENDENT GENE SILENCING1 gene codes for an S-adenosyl-L-homocysteine hydrolase required for DNA methylation-dependent gene silencing. Plant Cell 2005, 17, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J.-K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef]
- Gehring, M.; Huh, J.H.; Hsieh, T.-F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Galisteo, A.P.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008, 67, 671–681. [Google Scholar] [CrossRef]
- Agius, F.; Kapoor, A.; Zhu, J.-K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. USA 2006, 103, 11796–11801. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Chevala, V.V.S.N.; Shankar, R.; Jain, M. Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci. Rep. 2015, 5, 14922. [Google Scholar] [CrossRef]
- Popova, O.V.; Dinh, H.Q.; Aufsatz, W.; Jonak, C. The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol. Plant. 2013, 6, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, A.; Yabuta, Y.; Yoshida, E.; Maruta, T.; Yoshimura, K.; Shigeoka, S. Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J. 2006, 48, 535–547. [Google Scholar] [CrossRef]
- Swindell, W.R.; Huebner, M.; Weber, A.P. Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom. 2007, 8, 125. [Google Scholar] [CrossRef]
- Dolzhenko, E.; Smith, A.D. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinform. 2014, 15, 215. [Google Scholar] [CrossRef]
- Merchante, C.; Stepanova, A.N.; Alonso, J.M. Translation regulation in plants: An interesting past, an exciting present and a promising future. Plant J. 2017, 90, 628–653. [Google Scholar] [CrossRef] [PubMed]
- Artur, M.A.S.; Zhao, T.; Ligterink, W.; Schranz, E.; Hilhorst, H.W.M. Dissecting the genomic diversification of LATE EMBRYOGENESIS ABUNDANT (LEA) protein gene families in plants. Genome Biol. Evol. 2018, 11, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K. ABA signaling in stress-response and seed development. Plant Cell Rep. 2013, 32, 959–970. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2015, 44, D481–D487. [Google Scholar] [CrossRef]
- Goetz, A.E.; Wilkinson, M. Stress and the nonsense-mediated RNA decay pathway. Cell Mol. Life Sci. 2017, 74, 3509–3531. [Google Scholar] [CrossRef]
- Olvera-Carrillo, Y.; Campos, F.; Reyes, J.L.; Garciarrubio, A.; Covarrubias, A.A. Functional analysis of the group 4 late embryogenesis abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 2010, 154, 373–390. [Google Scholar] [CrossRef]
- Vishwakarma, K.; Upadhyay, N.; Kumar, N.; Yadav, G.; Singh, J.; Mishra, R.K.; Kumar, V.; Verma, R.; Upadhyay, R.G.; Pandey, M.; et al. Abscisic acid signaling and abiotic stress tolerance in plants: A review on current knowledge and future prospects. Front. Plant Sci. 2017, 8, 161. [Google Scholar] [CrossRef]
- Larkindale, J.; Knight, M.R. Protection against heat stress-induced oxidative damage in Arabidopsis involves calcium, abscisic acid, ethylene, and salicylic acid. Plant Physiol. 2002, 128, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, F.; Thomas, C.L.; Lederer, C.; Niu, Y.; Wang, D.; Maule, A.J. Virus induction of heat shock protein 70 reflects a general response to protein accumulation in the plant cytosol. Plant Physiol. 2005, 138, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Noël, L.D.; Cagna, G.; Stuttmann, J.; Wirthmüller, L.; Betsuyaku, S.; Witte, C.-P.; Bhat, R.; Pochon, N.; Colby, T.; Parker, J.E. Interaction between SGT1 and cytosolic/nuclear HSC70 chaperones regulates Arabidopsis immune responses. Plant Cell 2007, 19, 4061–4076. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.S.; Cho, E.J.; Choi, E.-Y.; Park, O.K. Analysis of the Arabidopsis nuclear proteome and its response to cold stress. Plant J. 2003, 36, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Hubert, D.A.; He, Y.; McNulty, B.C.; Tornero, P.; Dangl, J.L. Specific Arabidopsis HSP90. 2 alleles recapitulate RAR1 cochaperone function in plant NB-LRR disease resistance protein regulation. Proc. Natl. Acad Sci. USA 2009, 106, 9556–9563. [Google Scholar] [CrossRef]
- Takahashi, A.; Casais, C.; Ichimura, K.; Shirasu, K. HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc. Natl. Acad Sci. USA 2003, 100, 11777–11782. [Google Scholar] [CrossRef]
- Larkindale, J.; Hall, J.D.; Knight, M.R.; Vierling, E. Heat stress phenotypes of Arabidopsis mutants implicate multiple signaling pathways in the acquisition of thermotolerance. Plant Physiol. 2005, 138, 882–897. [Google Scholar] [CrossRef]
- Sharma, S.; Villamor, J.G.; Verslues, P.E. Essential Role of Tissue-Specific Proline Synthesis and Catabolism in Growth and Redox Balance at Low Water Potential. Plant Physiol. 2011, 157, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Parasuraman, B.; Laxmi, A.; Chattopadhyay, D. CIPK6, a CBL-interacting protein kinase is required for development and salt tolerance in plants. Plant J. 2009, 58, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernández, A.A.; Ortega-Amaro, M.A.; Delgado-Sánchez, P.; Salinas, J.; Jiménez-Bremont, J.F. AtGRDP1 Gene encoding a glycine-rich domain protein is involved in germination and responds to ABA signalling. Plant Mol. Biol. Rep. 2014, 32, 1187–1202. [Google Scholar] [CrossRef]
- Sarry, J.-E.; Kuhn, L.; Ducruix, C.; Lafaye, A.; Junot, C.; Hugouvieux, V.; Jourdain, A.; Bastien, O.; Fievet, J.B.; Vailhen, D.; et al. The early responses of Arabidopsis thaliana cells to cadmium exposure explored by protein and metabolite profiling analyses. Proteomics 2006, 6, 2180–2198. [Google Scholar] [CrossRef]
- Jones, A.M.E.; Thomas, V.; Bennett, M.H.; Mansfield, J.; Grant, M. Modifications to the Arabidopsis defense proteome occur prior to significant transcriptional change in response to inoculation with Pseudomonas syringae. Plant Physiol. 2006, 142, 1603–1620. [Google Scholar] [CrossRef]
- Paul, A.; Dasgupta, P.; Roy, D.; Chaudhuri, S. Comparative analysis of Histone modifications and DNA methylation at OsBZ8 locus under salinity stress in IR64 and Nonabokra rice varieties. Plant Mol. Biol. 2017, 95, 63–88. [Google Scholar] [CrossRef] [PubMed]
- Yaish, M.W.; Al-Lawati, A.; Al-Harrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, X.; Wang, H.; Shi, F.; Liu, Y.; Liu, J.; Li, L.; Wang, D.; Liu, B. Cytosine methylation alteration in natural populations of Leymus chinensis induced by multiple abiotic stresses. PLoS ONE 2013, 8, e55772. [Google Scholar] [CrossRef]
- Fan, H.H.; Wei, J.; Li, T.C.; Li, Z.P.; Guo, N.; Cai, Y.P.; Lin, Y. DNA methylation alterations of upland cotton (Gossypium hirsutum) in response to cold stress. Acta Physiol. Plant. 2013, 35, 2445–2453. [Google Scholar] [CrossRef]
- Shan, X.; Wang, X.; Yang, G.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Yuan, Y. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J. Plant Biol. 2013, 56, 32–38. [Google Scholar] [CrossRef]
- Ci, D.; Song, Y.; Tian, M.; Zhang, D. Methylation of miRNA genes in the response to temperature stress in Populus simonii. Front. Plant Sci. 2015, 6, 921. [Google Scholar] [CrossRef]
- Chwialkowska, K.; Nowakowska, U.; Mroziewicz, A.; Szarejko, I.; Kwasniewski, M. Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (Hordeum vulgare L.). J. Exp. Bot. 2016, 67, 1109–1121. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Surdonja, K.; Eggert, K.; Hajirezaei, M.-R.; Harshavardhan, V.; Seiler, C.; von Wirén, N.; Sreenivasulu, N.; Kuhlmann, M. Increase of DNA Methylation at the HvCKX2. 1 promoter by terminal drought stress in Barley. Epigenomes 2017, 1, 9. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Sun, M.; Zhang, T.; Li, H.; Chen, B.; Xu, K.; Gao, G.; Li, F.; Yan, G.; et al. Global DNA methylation variations after short-term heat shock treatment in cultured microspores of Brassica napus cv. Topas. Sci. Rep. 2016, 6, 38401. [Google Scholar] [CrossRef]
- Min, L.; Li, Y.; Hu, Q.; Zhu, L.; Gao, W.; Wu, Y.; Ding, Y.; Liu, S.; Yang, X.; Zhang, X. Sugar and auxin signaling pathways respond to high-temperature stress during anther development as revealed by transcript profiling analysis in cotton. Plant Physiol. 2014, 164, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins Jr, F.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Li, Y.; Kumar, S.; Qian, W. Active DNA demethylation: Mechanism and role in plant development. Plant Cell Rep. 2018, 37, 77–85. [Google Scholar] [CrossRef]
- Qu, A.-L.; Ding, Y.-F.; Jiang, Q.; Zhu, C. Molecular mechanisms of the plant heat stress response. Biochem. Biophys. Res. Commun. 2013, 432, 203–207. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.-D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- Feder, M.E.; Hofmann, G.E. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef]
- Panaretou, B.; Zhai, C. The heat shock proteins: Their roles as multi-component machines for protein folding. Fungal Biol. Rev. 2008, 22, 110–119. [Google Scholar] [CrossRef]
- Hu, W.; Hu, G.; Han, B. Genome-wide survey and expression profiling of heat shock proteins and heat shock factors revealed overlapped and stress specific response under abiotic stresses in rice. Plant Sci. 2009, 176, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Gallie, D.R.; Caldwell, C.; Pitto, L. Heat shock disrupts cap and poly (A) tail function during translation and increases mRNA stability of introduced reporter mRNA. Plant Physiol. 1995, 108, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Ishibashi, Y.; Shinmyo, A.; Kanaya, S.; Kato, K. Genome-wide analyses of early translational responses to elevated temperature and high salinity in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Yángüez, E.; Castro-Sanz, A.B.; Fernandez-Bautista, N.; Oliveros, J.C.; Castellano, M.M. Analysis of genome-wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PLoS ONE 2013, 8, e71425. [Google Scholar] [CrossRef] [PubMed]
- Merret, R.; Nagarajan, V.K.; Carpentier, M.-C.; Park, S.; Favory, J.-J.; Descombin, J.; Picart, C.; Charng, Y.; Green, P.J.; Deragon, J.-M.; et al. Heat-induced ribosome pausing triggers mRNA co-translational decay in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 4121–4132. [Google Scholar] [CrossRef] [PubMed]
- Karam, R.; Lou, C.-H.; Kroeger, H.; Huang, L.; Lin, J.H.; Wilkinson, M.F. The unfolded protein response is shaped by the NMD pathway. Embo Rep. 2015, 16, 599–609. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, B.; Harris, N.S.; Deyholos, M.K. Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 2007, 58, 3591–3607. [Google Scholar] [CrossRef]
- Kilian, J.; Whitehead, D.; Horak, J.; Wanke, D.; Weinl, S.; Batistic, O.; D’angelo, C.; Bornberg-Bauer, E.; Kudla, J.; Harter, K. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007, 50, 347–363. [Google Scholar] [CrossRef]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An Electronic Fluorescent Pictograph browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Goulas, E.; Schubert, M.; Kieselbach, T.; Kleczkowski, L.A.; Gardeström, P.; Schröder, W.; Hurry, V. The chloroplast lumen and stromal proteomes of Arabidopsis thaliana show differential sensitivity to short-and long-term exposure to low temperature. Plant J. 2006, 47, 720–734. [Google Scholar] [CrossRef]
- Richards, K.D.; Schott, E.J.; Sharma, Y.K.; Davis, K.R.; Gardner, R.C. Aluminum induces oxidative stress genes in Arabidopsis thaliana. Plant Physiol. 1998, 116, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Stancheva, I. Caught in conspiracy: Cooperation between DNA methylation and histone H3K9 methylation in the establishment and maintenance of heterochromatin. Biochem. Cell Biol. 2005, 83, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. Rev. Mutat. Res. 2008, 659, 40–48. [Google Scholar]
- Lauria, M.; Rossi, V. Epigenetic control of gene regulation in plants. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2011, 1809, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Terol, J.; Castillo, M.C.; Bargues, M.; Pérez-Alonso, M.; de Frutos, R. Structural and evolutionary analysis of the copia-like elements in the Arabidopsis thaliana genome. Mol. Biol. Evol. 2001, 18, 882–892. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2015. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 15 December 2020).
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Function | Gene ID | Gene Name | Gene Description | References |
|---|---|---|---|---|
| Protein folding, response to heat | AT3G12580 | HSP70 | Heat shock protein 70 | [39] |
| AT5G02500 | HSP70-1 | Heat shock protein 70-1 | [40] | |
| AT4G24280 | HSP70-6 | Heat shock protein 70-6 | [41] | |
| AT5G56030 | HSP81-2 | Heat shock protein 81-2 | [42] | |
| AT5G52640 | HSP90-1 | Heat shock protein 90-1 | [43] | |
| AT1G74310 | HSP101 | Heat shock protein 101 | [44] | |
| Regulation of translation | AT3G49910 | RPL26A | 60S ribosomal protein L26-1 | [41] |
| AT2G37190 | RPL12A | 60S ribosomal protein L12-1 | [41] | |
| Response to water deprivation | AT5G06760 | LEA46 | Late embryogenesis abundant protein 46 | [36] |
| AT3G30775 | POX1 | Proline dehydrogenase 1 | [45] | |
| AT4G30960 | CIPK6 | CBL-interacting serine/threonine-protein kinase 6 | [46] | |
| ABA signaling pathway | AT2G22660 | GRDP1 | Glycine-rich domain-containing protein 1 | [47] |
| Oxidation-reduction process | AT5G14780 | FDH1 | Formate dehydrogenase | [48] |
| AT5G66570 | PSBO1 | Oxygen-evolving enhancer protein 1-1 | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korotko, U.; Chwiałkowska, K.; Sańko-Sawczenko, I.; Kwasniewski, M. DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 1555. https://doi.org/10.3390/ijms22041555
Korotko U, Chwiałkowska K, Sańko-Sawczenko I, Kwasniewski M. DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana. International Journal of Molecular Sciences. 2021; 22(4):1555. https://doi.org/10.3390/ijms22041555
Chicago/Turabian StyleKorotko, Urszula, Karolina Chwiałkowska, Izabela Sańko-Sawczenko, and Miroslaw Kwasniewski. 2021. "DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana" International Journal of Molecular Sciences 22, no. 4: 1555. https://doi.org/10.3390/ijms22041555
APA StyleKorotko, U., Chwiałkowska, K., Sańko-Sawczenko, I., & Kwasniewski, M. (2021). DNA Demethylation in Response to Heat Stress in Arabidopsis thaliana. International Journal of Molecular Sciences, 22(4), 1555. https://doi.org/10.3390/ijms22041555