
Bcl-xL as a Modulator of Senescence and Aging




Abstract
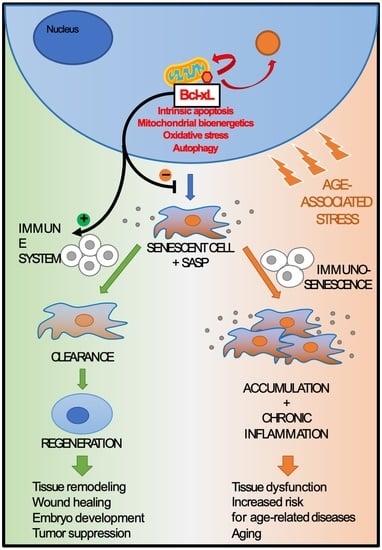
1. Introduction
2. Measuring Cellular Senescence In Vivo
3. Senescence: From Development to Adulthood
4. Senescence Role in Ordinary Aging
4.1. Senescence and Age-Related Diseases
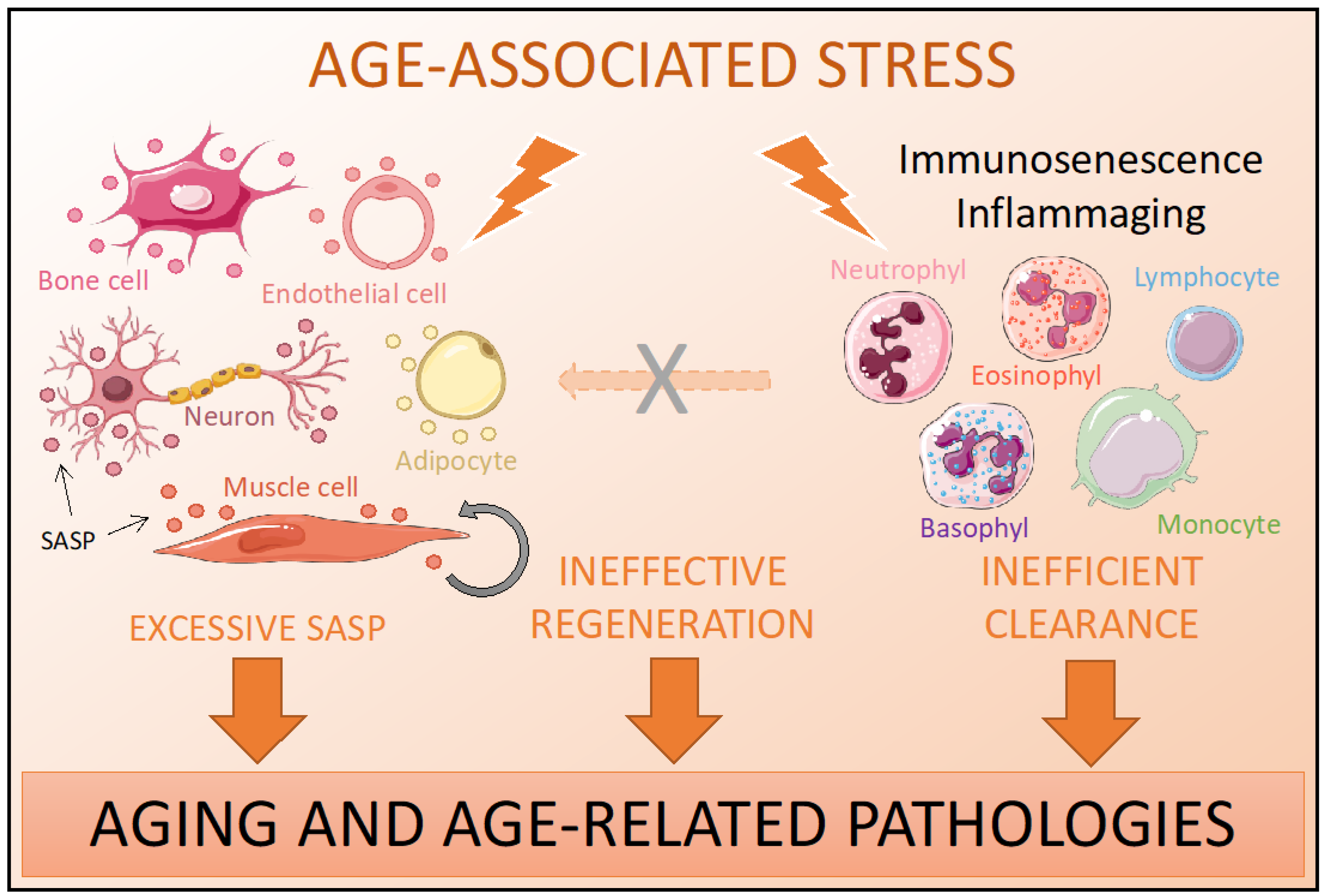
4.2. Immunosenescence and Inflammaging
5. Senescence Role in Exceptional Aging
5.1. Balance between Senescence and Apoptosis in Exceptional Aging
5.2. Bcl-xL Modulates Senescence in Exceptional Aging
5.3. Bcl-xL Maintains Immunosurveillance of Senescent Cells in Exceptional Aging
6. Bcl-xL as a Senolytic
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| γH2AX | Gamma H2A histone family member X |
| ATM | Ataxia Telangiectasia Mutated |
| Bcl-xL | B-cell lymphoma extra-large |
| DDR | DNA damage response |
| DISC | Death-inducing signaling complex |
| FADD | Fas-associated protein with death domain |
| GATA4 | GATA binding protein 4 |
| HMGB1 | High mobility group box 1 |
| HSC | Hematopoietic stem cells |
| HUVEC | Human umbilical vein endothelial cells |
| IFNG | Interferon- γ |
| IKK/NF-κB | IK kinase/Nuclear factor κB |
| IL | Interleukins |
| iPSC | Induced pluripotent stem cell |
| JAK/STAT | Janus kinase/Signal transducer and activator of transcription |
| MEFs | Mouse embryonic fibroblasts |
| MMPs | Matrix metalloproteinases |
| NK | Natural Killer |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PBMC | Peripheral blood mononuclear cells |
| PI3K | Phospho-Inositol 3 Kinase |
| ROS | Reactive oxygen species |
| SA-β-gal | Senescence associated- β-galactosidase |
| SADS | Senescence associated- distension of satellite DNA |
| SAHF | Senescence associated- heterochromatin foci |
| SASP | Senescence-associated secretory phenotype |
| SP1 | SP1 transcription factor |
| TAF | Telomere associated- foci |
| TCR | T-cell receptor |
| TGF β1 | Transforming growth factor- β1 |
| TNF | Tumor necrosis factor |
References
- Griffin, J.P. Changing life expectancy throughout history. J. R Soc. Med. 2008, 101, 577. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Demographics Collaborators. Global age-sex-specific fertility, mortality, healthy life expectancy (HALE), and population estimates in 204 countries and territories, 1950–2019: A comprehensive demographic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1160–1203. [Google Scholar] [CrossRef]
- Olshansky, S.J. From Lifespan to Healthspan. JAMA 2018, 320, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T.; Wan, T.; Miwa, S. Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxid Redox Signal 2021, 34, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, M.; Matheu, A. Autophagy in stem cell aging. Aging Cell 2017, 16, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging--when “all you can eat” is yourself. Sci. Aging Knowl. Env. 2003, 2003, pe25. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining” clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Galluzzi, L.; Tajeddine, N.; Ortiz, C.; Criollo, A.; Tasdemir, E.; Morselli, E.; Ben Younes, A.; Maiuri, M.C.; Lavandero, S.; et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path—A mini-review. Gerontology 2008, 54, 92–99. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharm. Toxicol. 2020. [Google Scholar] [CrossRef]
- Mohamad Kamal, N.S.; Safuan, S.; Shamsuddin, S.; Foroozandeh, P. Aging of the cells: Insight into cellular senescence and detection Methods. Eur. J. Cell Biol. 2020, 99, 151108. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Kouroupi, M.; Balaska, K.; Koukourakis, M.I. Immunohistochemical detection of senescence markers in human sarcomas. Pathol. Res. Pr. 2020, 216, 152800. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Nakabayashi, K.; Fujii, M.; Scherer, S.W.; Ayusawa, D. Early BrdU-responsive genes constitute a novel class of senescence-associated genes in human cells. Exp. Cell Res. 2005, 304, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Narita, M. Cellular senescence and chromatin organisation. Br. J. Cancer 2007, 96, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.P.; Campeau, E.; Beauséjour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Lee, J.J.; Park, I.H.; Rhee, W.J.; Kim, H.S.; Shin, J.S. HMGB1 modulates the balance between senescence and apoptosis in response to genotoxic stress. FASEB J. 2019, 33, 10942–10953. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Bernards, R. Senescence, wound healing and cancer: The PAI-1 connection. Cell Cycle 2006, 5, 2697–2703. [Google Scholar] [CrossRef] [PubMed]
- Rochette, P.J.; Brash, D.E. Progressive apoptosis resistance prior to senescence and control by the anti-apoptotic protein BCL-xL. Mech Ageing Dev. 2008, 129, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Jonchère, B.; Vétillard, A.; Toutain, B.; Lam, D.; Bernard, A.C.; Henry, C.; De Carné Trécesson, S.; Gamelin, E.; Juin, P.; Guette, C.; et al. Irinotecan treatment and senescence failure promote the emergence of more transformed and invasive cells that depend on anti-apoptotic Mcl-1. Oncotarget 2015, 6, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.L.; Hoffmann, R.; González-López, C.; Doherty, G.J.; Korkola, J.E.; Muñoz-Espín, D. Cellular senescence in cancer: From mechanisms to detection. Mol. Oncol. 2020. [Google Scholar] [CrossRef]
- Gasmi, A.; Chirumbolo, S.; Peana, M.; Mujawdiya, P.K.; Dadar, M.; Menzel, A.; Bjørklund, G. Biomarkers of senescence during aging as possible warnings to use preventive measures. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Pedraza-Vázquez, G.; Maciel-Barón, L.A.; Königsberg, M. Reflections on the role of senescence during development and aging. Arch. Biochem. Biophys. 2016, 598, 40–49. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Hampel, B.; Malisan, F.; Niederegger, H.; Testi, R.; Jansen-Dürr, P. Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 2004, 39, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Mausset-Bonnefont, A.L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguín, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407–414.e404. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 2016, 8, 1294–1315. [Google Scholar] [CrossRef]
- von Kobbe, C. Cellular senescence: A view throughout organismal life. Cell Mol. Life Sci. 2018, 75, 3553–3567. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Zingoni, A.; Soriani, A.; Santoni, A. Senescent cells: Living or dying is a matter of NK cells. J. Leukoc. Biol. 2019, 105, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Borrás, C.; Miquel, J. Theories of ageing. IUBMB Life 2007, 59, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Senolytics and Senostatics: A Two-Pronged Approach to Target Cellular Senescence for Delaying Aging and Age-Related Diseases. Mol. Cells 2019, 42, 821–827. [Google Scholar] [CrossRef]
- von Zglinicki, T.; Saretzki, G.; Docke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Vina-Almunia, J.; Ingles, M.; Sanz-Ros, J.; Gambini, J.; Ibanez-Cabellos, J.S.; Garcia-Gimenez, J.L.; Vina, J.; Borras, C. Role of p16INK4a and BMI-1 in oxidative stress-induced premature senescence in human dental pulp stem cells. Redox Biol. 2017, 12, 690–698. [Google Scholar] [CrossRef]
- Vono, R.; Jover Garcia, E.; Spinetti, G.; Madeddu, P. Oxidative Stress in Mesenchymal Stem Cell Senescence: Regulation by Coding and Noncoding RNAs. Antioxid. Redox Signal. 2018, 29, 864–879. [Google Scholar] [CrossRef]
- Moon, K.C.; Yang, J.P.; Lee, J.S.; Jeong, S.H.; Dhong, E.S.; Han, S.K. Effects of Ultraviolet Irradiation on Cellular Senescence in Keratinocytes Versus Fibroblasts. J. Craniofac. Surg. 2019, 30, 270–275. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends. Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging. Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005, 48, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Markowski, D.N.; Thies, H.W.; Gottlieb, A.; Wenk, H.; Wischnewsky, M.; Bullerdiek, J. HMGA2 expression in white adipose tissue linking cellular senescence with diabetes. Genes Nutr. 2013, 8, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, M.J.; Schlauch, K.; Lenburg, M.E.; Tchkonia, T.; Pirtskhalava, T.; Cartwright, A.; Thomou, T.; Kirkland, J.L. Aging, depot origin, and preadipocyte gene expression. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Gustafson, B.; Kirkland, J.L.; Smith, U. Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 2019, 62, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Min. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Chinta, S.J.; Lieu, C.A.; Demaria, M.; Laberge, R.M.; Campisi, J.; Andersen, J.K. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to Parkinson’s disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- McEwen, J.E.; Zimniak, P.; Mehta, J.L.; Shmookler Reis, R.J. Molecular pathology of aging and its implications for senescent coronary atherosclerosis. Curr. Opin. Cardiol. 2005, 20, 399–406. [Google Scholar] [CrossRef]
- Burton, D.G.; Matsubara, H.; Ikeda, K. Pathophysiology of vascular calcification: Pivotal role of cellular senescence in vascular smooth muscle cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence-Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Santoro, A.; Capri, M. The complex relationship between Immunosenescence and Inflammaging: Special issue on the New Biomedical Perspectives. Semin. Immunopathol. 2020, 42, 517–520. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Du, J.; Klein, J.D.; Hassounah, F.; Zhang, J.; Zhang, C.; Wang, X.H. Aging increases CCN1 expression leading to muscle senescence. Am. J. Physiol. Cell Physiol. 2014, 306, C28–C36. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, J.; Spee, C.; Ryan, S.J.; Hinton, D.R. BMP4 mediates oxidative stress-induced retinal pigment epithelial cell senescence and is overexpressed in age-related macular degeneration. J. Biol. Chem. 2009, 284, 9529–9539. [Google Scholar] [CrossRef]
- Le Maitre, C.L.; Freemont, A.J.; Hoyland, J.A. Accelerated cellular senescence in degenerate intervertebral discs: A possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res. 2007, 9, R45. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- Zampino, M.; Ferrucci, L.; Semba, R.D. Biomarkers in the path from cellular senescence to frailty. Exp. Gerontol. 2020, 129, 110750. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.; Horvath, S.; Raj, K. Epigenetic clock analyses of cellular senescence and ageing. Oncotarget 2016, 7, 8524–8531. [Google Scholar] [CrossRef]
- Pawelec, G.; Adibzadeh, M.; Pohla, H.; Schaudt, K. Immunosenescence: Ageing of the immune system. Immunol. Today 1995, 16, 420–422. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug. Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef]
- Attaallah, A.; Lenzi, M.; Marchionni, S.; Bincoletto, G.; Cocchi, V.; Croco, E.; Hrelia, P.; Hrelia, S.; Sell, C.; Lorenzini, A. A pro longevity role for cellular senescence. Geroscience 2020, 42, 867–879. [Google Scholar] [CrossRef]
- Trougakos, I.P.; Petropoulou, C.; Franceschi, C.; Gonos, E.S. Reduced expression levels of the senescence biomarker clusterin/apolipoprotein j in lymphocytes from healthy centenarians. Ann. N. Y. Acad. Sci. 2006, 1067, 294–300. [Google Scholar] [CrossRef]
- Borras, C.; Abdelaziz, K.M.; Gambini, J.; Serna, E.; Ingles, M.; de la Fuente, M.; Garcia, I.; Matheu, A.; Sanchis, P.; Belenguer, A.; et al. Human exceptional longevity: Transcriptome from centenarians is distinct from septuagenarians and reveals a role of Bcl-xL in successful aging. Aging (Albany NY) 2016, 8, 3185–3208. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Monti, D.; Franceschi, C. The immune system in the elderly: Activation-induced and damage-induced apoptosis. Immunol. Res. 2004, 30, 81–94. [Google Scholar] [CrossRef]
- Borrás, C.; Mas-Bargues, C.; Román-Domínguez, A.; Sanz-Ros, J.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. BCL-xL, a Mitochondrial Protein Involved in Successful Aging: From, C. elegans to Human Centenarians. Int. J. Mol. Sci. 2020, 21, 418. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.A.; Porter, G.A.; Alavian, K.N. Bcl-xL in neuroprotection and plasticity. Front. Physiol. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Scheckhuber, C.Q.; Erjavec, N.; Tinazli, A.; Hamann, A.; Nyström, T.; Osiewacz, H.D. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat. Cell Biol. 2007, 9, 99–105. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef]
- Ni, L.; Li, T.; Liu, B.; Song, X.; Yang, G.; Wang, L.; Miao, S.; Liu, C. The protective effect of Bcl-xl overexpression against oxidative stress-induced vascular endothelial cell injury and the role of the Akt/eNOS pathway. Int. J. Mol. Sci. 2013, 14, 22149–22162. [Google Scholar] [CrossRef]
- Bandiera, S.; Matégot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19. [Google Scholar] [CrossRef]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging (Albany NY) 2018, 10, 2855–2873. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.G.; Dong, J.M.; Manser, E.; Song, H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J. Biol. Chem. 2008, 283, 26274–26282. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef]
- Burton, D.G.A.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Malavolta, M. NK and NKT cell functions in immunosenescence. Aging Cell 2004, 3, 177–184. [Google Scholar] [CrossRef]
- Campos, C.; Pera, A.; Lopez-Fernandez, I.; Alonso, C.; Tarazona, R.; Solana, R. Proinflammatory status influences NK cells subsets in the elderly. Immunol. Lett. 2014, 162, 298–302. [Google Scholar] [CrossRef]
- Peeper, D.S. Ageing: Old cells under attack. Nature 2011, 479, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Korsmeyer, S.J. Apoptosis in the development and maintenance of the immune system. Nat. Immunol. 2003, 4, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, D.; He, J.; Chen, L.; Li, H. Bcl-X(L): A multifunctional anti-apoptotic protein. Pharm. Res. 2020, 151, 104547. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, J.; Zeng, H.; Shao, L. Senescent Cell Depletion Through Targeting BCL-Family Proteins and Mitochondria. Front. Physiol. 2020, 11, 593630. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Mak, S.S.; Moriyama, M.; Nishioka, E.; Osawa, M.; Nishikawa, S. Indispensable role of Bcl2 in the development of the melanocyte stem cell. Dev. Biol. 2006, 291, 144–153. [Google Scholar] [CrossRef]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.M. Reversibility of cellular aging by reprogramming through an embryonic-like state: A new paradigm for human cell rejuvenation. Cent. Asian J. Glob. Health 2013, 2, 88. [Google Scholar] [CrossRef] [PubMed]
- Su, R.J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient generation of integration-free ips cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef]
- Giacconi, R.; Malavolta, M.; Costarelli, L.; Provinciali, M. Cellular Senescence and Inflammatory Burden as Determinants of Mortality in Elderly People Until the Extreme old age. EBioMedicine 2015, 2, 1316–1317. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Marker | Senescence | Ref. |
|---|---|---|
| Proliferation (Ki67, BrdU assay) | Absent | [15,16] |
| Cell cycle inhibitors (p21CIP1, p16INK4a) | Increased | [17,18,19] |
| DNA damage (p53, γH2AX foci, TAF, SAHF, SADF) | Increased | [20,21,22,23,24,25] |
| SA-β-gal activity | Increased | [26,27,28] |
| Lamin B1 | Decreased | [29] |
| Nuclear exclusion of HMGB1 | Present | [30] |
| SASP factors (IL-1, IL-6, IL-8, PAI-1, MMPs) | Present | [31,32] |
| Anti-apoptotic proteins (Bcl-xL, MCL-1) | Increased | [33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mas-Bargues, C.; Borrás, C.; Viña, J. Bcl-xL as a Modulator of Senescence and Aging. Int. J. Mol. Sci. 2021, 22, 1527. https://doi.org/10.3390/ijms22041527
Mas-Bargues C, Borrás C, Viña J. Bcl-xL as a Modulator of Senescence and Aging. International Journal of Molecular Sciences. 2021; 22(4):1527. https://doi.org/10.3390/ijms22041527
Chicago/Turabian StyleMas-Bargues, Cristina, Consuelo Borrás, and Jose Viña. 2021. "Bcl-xL as a Modulator of Senescence and Aging" International Journal of Molecular Sciences 22, no. 4: 1527. https://doi.org/10.3390/ijms22041527
APA StyleMas-Bargues, C., Borrás, C., & Viña, J. (2021). Bcl-xL as a Modulator of Senescence and Aging. International Journal of Molecular Sciences, 22(4), 1527. https://doi.org/10.3390/ijms22041527