MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
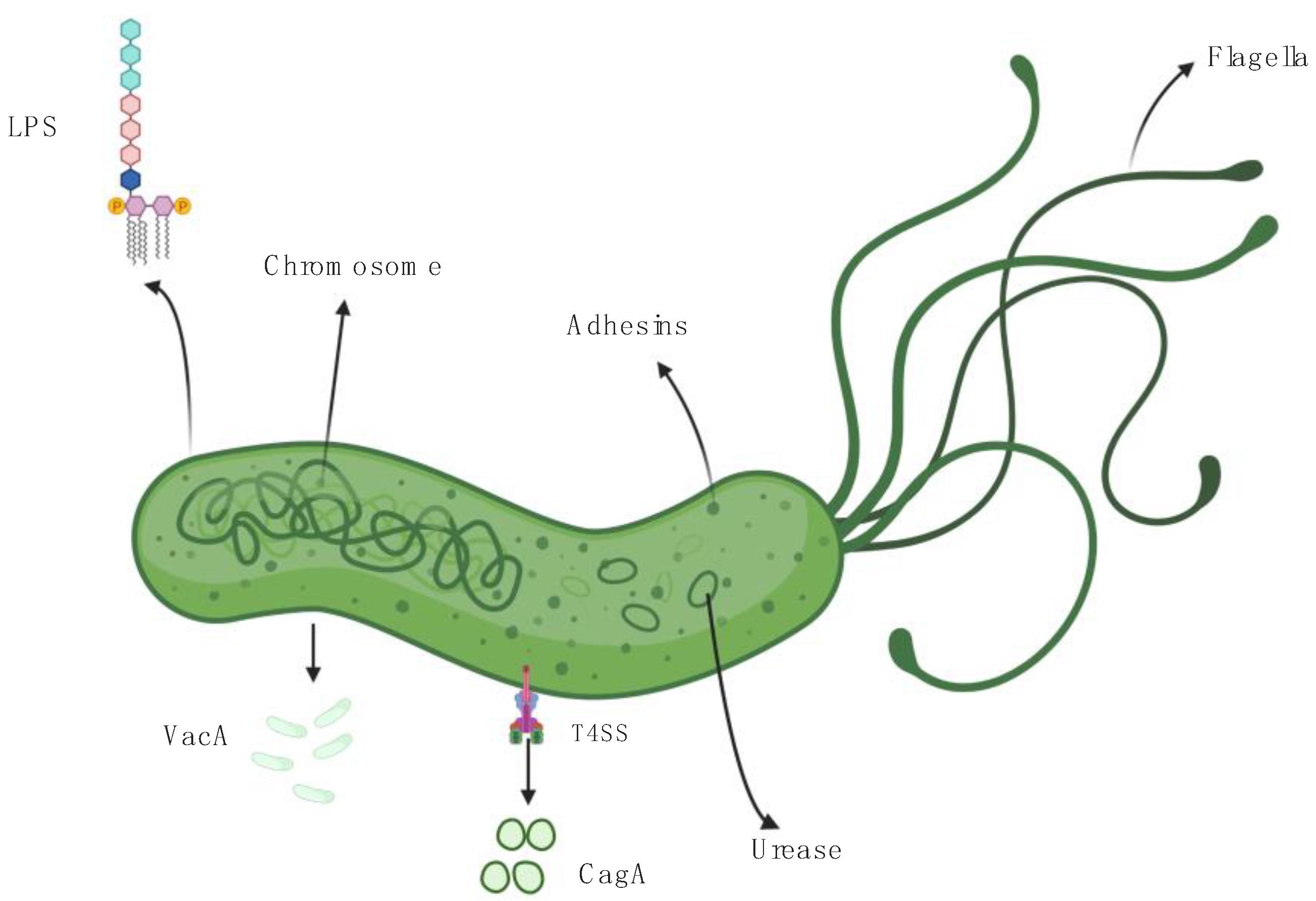
2. Helicobacter pylori, Virulence Factors, and Host Immune Response
3. Helicobacter pylori—Driven Pathways of Gastric Carcinogenesis
4. MiRNAs as Modulators of Innate Immune Response and H. pylori Associated Inflammation
4.1. Let-7 Family of miRNAs
4.2. MiRNA-155
4.3. MiRNA-146
4.4. MiRNA-125
4.5. MiRNA-21
4.6. MiRNA-223
4.7. Other miRNAs and H. pylori
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activator protein 1 |
| ARID1A | AT-rich interacting domain containing protein 1A |
| BRMS1 | breast cancer metastasis suppressor 1 |
| CagA | cytotoxin-associated gene A |
| CAMP | cationic antimicrobial peptide |
| EMT | Epithelial–mesenchymal transition |
| GRO | growth-related oncogene |
| H. pylori | Helicobacter pylori |
| HP-NAP | Helicobacter pylori neutrophil-activating protein |
| ICAM-1 | intercellular adhesion molecule-1 |
| IFNγ | interferon γ |
| IL | interleukin |
| IRAK1 | IL-1 receptor associated kinase 1 |
| Let-7 | lethal 7 |
| LPS | lipopolysaccharide |
| MALT | mucosa-associated lymphoid tissue |
| MAPK | mitogen-activated protein kinase |
| MHC | major histocompatibility complex |
| MIP | macrophage inflammatory protein |
| miRNA | microRNA |
| mRNA | messenger RNA |
| NF-kB | nuclear factor-kappa B |
| NOX1 | neutrophil NADPH oxidase |
| PAMP | pathogen-associated molecular pattern |
| PDCD4 | programmed cell death protein 4 |
| PRM | pattern-recognition molecule |
| ROS | reactive oxygen species |
| T4SS | type IV secretion system |
| Th | T helper cells |
| TLR | toll-like receptor |
| TRAF6 | tumor necrosis factor receptor associated factor-6 |
| TNF | tumor necrosis factor |
| UTR | untranslated region |
| VacA | vacuolating cytotoxin |
| VEGF | vascular endothelial growth factor |
References
- Herrera, V.; Parsonnet, J. Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 2009, 15, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.R.; Whitmire, J.M.; Merrell, D.S. A Tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front. Microbiol. 2010, 1, 115. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, A.R. Helicobacter, inflammation, and gastric cancer. Curr. Pathobiol. Rep. 2013, 1, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Toh, J.W.T.; Wilson, R.B. Pathways of Gastric Carcinogenesis, Helicobacter pylori virulence and interactions with antioxidant systems, vitamin C and phytochemicals. Int. J. Mol. Sci. 2020, 21, 6451. [Google Scholar] [CrossRef]
- Chiba, T.; Marusawa, H.; Seno, H.; Watanabe, N. Mechanism for gastric cancer development by Helicobacter pylori infection. J. Gastroenterol. Hepatol. 2008, 23, 1175–1181. [Google Scholar] [CrossRef]
- Ito, N.; Tsujimoto, H.; Ueno, H.; Xie, Q.; Shinomiya, N. Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer. J. Clin. Med. 2020, 9, 3699. [Google Scholar] [CrossRef]
- Krzyżek, P.; Grande, R.; Migdał, P.; Paluch, E.; Gościniak, G. Biofilm formation as a complex result of virulence and adaptive responses of Helicobacter pylori. Pathogens 2020, 9, 1062. [Google Scholar] [CrossRef]
- O’Connor, A.; Gisbert, J.P.; McNamara, D.; O’Morain, C. Treatment of Helicobacter pylori infection 2011. Helicobacter 2011, 16 (Suppl. S1), 53–58. [Google Scholar] [CrossRef]
- Ohkusa, T.; Fujiki, K.; Takashimizu, I.; Kumagai, J.; Tanizawa, T.; Eishi, Y.; Yokoyama, T.; Watanabe, M. Improvement in atrophic gastritis and intestinal metaplasia in patients in whom Helicobacter pylori was eradicated. Ann. Intern Med. 2001, 134, 380–386. [Google Scholar] [CrossRef]
- Liu, K.S.-H.; Wong, I.O.-L.; Leung, W.K. Helicobacter pylori associated gastric intestinal metaplasia: Treatment and surveillance. World J. Gastroenterol. 2016, 22, 1311–1320. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and Exosomal MicroRNA: Trafficking, sorting, and function. Genomics Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Baltimore, D. MicroRNAs and immunity: Tiny players in a big field. Immunity 2007, 26, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Tuschl, T. RISC Is a 5’ phosphomonoester-producing RNA endonuclease. Genes Dev. 2004, 18, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.M.; Mott, J.L. Overview of MicroRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of MicroRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Banzhaf-Strathmann, J.; Edbauer, D. Good Guy or Bad Guy: The opposing roles of MicroRNA 125b in cancer. Cell Commun. Signal 2014, 12, 30. [Google Scholar] [CrossRef]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release mechanisms, functions and challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. MicroRNA Regulation of Inflammatory Responses. Annu. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. MiR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Zabaleta, J. MicroRNA: A Bridge from H. pylori infection to gastritis and gastric cancer development. Front. Genet. 2012, 3, 294. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.C.; Blair, K.M.; Taylor, J.A.; Petersen, T.W.; Sessler, T.; Tull, C.M.; Leverich, C.K.; Collar, A.L.; Wyckoff, T.J.; Biboy, J.; et al. A Genome-wide Helicobacter pylori morphology screen uncovers a membrane-spanning helical cell shape complex. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R. Cell morphology as a virulence determinant: Lessons from Helicobacter pylori. Curr. Opin. Microbiol. 2020, 54, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Denic, M.; Touati, E.; De Reuse, H. Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2020, 25 (Suppl. S1), e12736. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowski, E.; Auger, R.; Tian, X.; Hicham, S.; Ecobichon, C.; Roure, S.; Douglass, M.V.; Trent, M.S.; Mengin-Lecreulx, D.; Touzé, T.; et al. HupA, the main undecaprenyl pyrophosphate and phosphatidylglycerol phosphate phosphatase in Helicobacter pylori is essential for colonization of the stomach. PLoS Pathog. 2019, 15, e1007972. [Google Scholar] [CrossRef]
- Scott, D.R.; Marcus, E.A.; Wen, Y.; Singh, S.; Feng, J.; Sachs, G. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with urei, a channel for urea and its metabolites, CO2, NH3, and NH4(+), is necessary for acid survival of Helicobacter pylori. J. Bacteriol. 2010, 192, 94–103. [Google Scholar] [CrossRef]
- Schoep, T.D.; Fulurija, A.; Good, F.; Lu, W.; Himbeck, R.P.; Schwan, C.; Choi, S.S.; Berg, D.E.; Mittl, P.R.E.; Benghezal, M.; et al. Surface properties of Helicobacter pylori urease complex are essential for persistence. PLoS ONE 2010, 5, e15042. [Google Scholar] [CrossRef]
- Lytton, S.D.; Fischer, W.; Nagel, W.; Haas, R.; Beck, F.X. Production of ammonium by Helicobacter pylori mediates occludin processing and disruption of tight junctions in Caco-2 cells. Microbiology 2005, 151, 3267–3276. [Google Scholar] [CrossRef]
- Fritz, J.H.; Le Bourhis, L.; Magalhaes, J.G.; Philpott, D.J. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol. 2008, 29, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Meliț, L.E.; Mărginean, C.O.; Mărginean, C.D.; Mărginean, M.O. The Relationship between Toll-like receptors and Helicobacter pylori-related gastropathies: Still a controversial topic. J. Immunol. Res. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Tsoporis, J.N.; Jia, S.-H.; dos Santos, C.C.; Parker, T.G.; Marshall, J.C. Toll-like receptors, associated biochemical signaling networks, and S100 Ligands. Shock 2020. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-KappaB by toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Bockerstett, K.A.; Osaki, L.H.; Petersen, C.P.; Cai, C.W.; Wong, C.F.; Nguyen, T.-L.M.; Ford, E.L.; Hoft, D.F.; Mills, J.C.; Goldenring, J.R.; et al. Interleukin-17A promotes parietal cell atrophy by inducing apoptosis. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 678–690.e1. [Google Scholar] [CrossRef]
- Amedei, A.; Cappon, A.; Codolo, G.; Cabrelle, A.; Polenghi, A.; Benagiano, M.; Tasca, E.; Azzurri, A.; D’Elios, M.M.; Del Prete, G.; et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J. Clin. Investig. 2006, 116, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; de Bernard, M. Molecular and cellular mechanisms of action of the vacuolating cytotoxin (VacA) and neutrophil-activating protein (HP-NAP) virulence factors of Helicobacter pylori. Microbes Infect. 2003, 5, 715–721. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhong, X.; Zheng, S.; Du, Q.; Xu, W. Transformed immortalized gastric epithelial cells by virulence factor CagA of Helicobacter pylori through Erk mitogen-activated protein kinase pathway. Oncogene 2005, 24, 3886–3895. [Google Scholar] [CrossRef]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef]
- Smolka, A.J.; Backert, S. How Helicobacter pylori infection controls gastric acid secretion. J. Gastroenterol. 2012, 47, 609–618. [Google Scholar] [CrossRef]
- Suzuki, M.; Mimuro, H.; Kiga, K.; Fukumatsu, M.; Ishijima, N.; Morikawa, H.; Nagai, S.; Koyasu, S.; Gilman, R.H.; Kersulyte, D.; et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host. Microbe 2009, 5, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori Activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol. 2009, 11, 70–82. [Google Scholar] [CrossRef]
- Supajatura, V.; Ushio, H.; Wada, A.; Yahiro, K.; Okumura, K.; Ogawa, H.; Hirayama, T.; Ra, C. Cutting Edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J. Immunol. 2002, 168, 2603–2607. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.-Y.; Jones, N.L. Helicobacter pylori Strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (Coronin 1) protein. Cell Microbiol. 2003, 5, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori virulence factors-mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells 2020, 10, 27. [Google Scholar] [CrossRef]
- Whitney, A.E.; Guarner, J.; Hutwagner, L.; Gold, B.D. Helicobacter pylori gastritis in children and adults: Comparative histopathologic study. Ann. Diagn. Pathol. 2000, 4, 279–285. [Google Scholar] [CrossRef]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and grading of gastritis. The updated Sydney system. International Workshop on the histopathology of gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef]
- Algood, H.M.S.; Gallo-Romero, J.; Wilson, K.T.; Peek, R.M.; Cover, T.L. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol. Med. Microbiol. 2007, 51, 577–586. [Google Scholar] [CrossRef]
- Xu, B.; Aoyama, K.; Takeuchi, M.; Matsushita, T.; Takeuchi, T. Expression of cytokine MRNAs in mice cutaneously exposed to formaldehyde. Immunol. Lett. 2002, 84, 49–55. [Google Scholar] [CrossRef]
- Carbo, A.; Bassaganya-Riera, J.; Pedragosa, M.; Viladomiu, M.; Marathe, M.; Eubank, S.; Wendelsdorf, K.; Bisset, K.; Hoops, S.; Deng, X.; et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS ONE 2013, 8, e73365. [Google Scholar] [CrossRef]
- Olokoba, A.B.; Obateru, O.A.; Bojuwoye, M.O. Helicobacter pylori Eradication therapy: A review of current trends. Niger. Med. J. 2013, 54, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Rumi, M.A.K.; Kadowaki, Y.; Ortega-Cava, C.F.; Yuki, T.; Yoshino, N.; Miyaoka, Y.; Kazumori, H.; Ishimura, N.; Amano, Y.; et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J. Immunol. 2004, 173, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Khoi, P.N.; Xia, Y.; Park, J.S.; Joo, Y.E.; Kim, K.K.; Choi, S.Y.; Jung, Y.D. Helicobacter pylori and interleukin-8 in gastric cancer. World J. Gastroenterol. 2013, 19, 8192–8202. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef]
- Chu, S.H.; Kim, H.; Seo, J.Y.; Lim, J.W.; Mukaida, N.; Kim, K.H. Role of NF-KappaB and AP-1 on helicobater pylori-induced IL-8 expression in AGS cells. Dig. Dis. Sci. 2003, 48, 257–265. [Google Scholar] [CrossRef]
- Chen, X.-M.; Splinter, P.L.; O’Hara, S.P.; LaRusso, N.F. A Cellular micro-RNA, Let-7i, regulates toll-like receptor 4 expression and contributes to cholangiocyte immune responses against cryptosporidium parvum infection. J. Biol. Chem. 2007, 282, 28929–28938. [Google Scholar] [CrossRef]
- Yu, H.; Zeng, J.; Liang, X.; Wang, W.; Zhou, Y.; Sun, Y.; Liu, S.; Li, W.; Chen, C.; Jia, J. Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4). PLoS ONE 2014, 9, e105306. [Google Scholar] [CrossRef]
- Abdullah, M.; Greenfield, L.K.; Bronte-Tinkew, D.; Capurro, M.I.; Rizzuti, D.; Jones, N.L. VacA promotes CagA accumulation in gastric epithelial cells during Helicobacter pylori infection. Sci. Rep. 2019, 9, 38. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Prattichizzo, F.; Babini, L.; Graciotti, L.; Recchioni, R.; Procopio, A.D. Toll like receptor signaling in “Inflammaging”: MicroRNA as new players. Immun. Ageing 2013, 10, 11. [Google Scholar] [CrossRef]
- Banerjee, S.; Thompson, W.E.; Chowdhury, I. Emerging roles of microRNAs in the regulation of toll-like receptor (TLR)-signaling. Front. Biosci 2021, 26, 771–796. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Hao, D.; Liu, X.; Wang, D.; Ning, N.; Li, X. Aberrant regulation of the LIN28A/LIN28B and Let-7 Loop in human malignant tumors and its effects on the hallmarks of Cancer. Mol. Cancer 2015, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Roush, S.; Slack, F.J. The Let-7 Family of microRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.; Wang, W.; Dai, Y.; Wang, S.; Chu, Y.; Li, J. Let-7b Is Involved in the Inflammation and immune responses associated with Helicobacter pylori infection by targeting toll-like receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H.; Matsushima, K.; Inoue, N.; Hayashi, T.; Nakayama, T.; Kunizaki, M.; Hidaka, S.; Nakayama, M.; Hisatsune, J.; Nakashima, M.; et al. Interweaving microRNAs and proinflammatory cytokines in gastric mucosa with reference to H. pylori infection. J. Clin. Immunol. 2012, 32, 290–299. [Google Scholar] [CrossRef]
- Matsushima, K.; Isomoto, H.; Inoue, N.; Nakayama, T.; Hayashi, T.; Nakayama, M.; Nakao, K.; Hirayama, T.; Kohno, S. MicroRNA signatures in Helicobacter pylori-infected gastric mucosa. Int. J. Cancer 2011, 128, 361–370. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Wang, X.-J.; Li, G.-X.; Yang, E.; Yang, N.-M. Detection of Let-7a microRNA by real-time PCR in gastric carcinoma. World J. Gastroenterol. 2007, 13, 2883–2888. [Google Scholar] [CrossRef]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.; Yamaguchi, K.; et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 2010, 5, e13247. [Google Scholar] [CrossRef]
- Staedel, C.; Darfeuille, F. MicroRNAs and bacterial infection. Cell. Microbiol. 2013, 15, 1496–1507. [Google Scholar] [CrossRef]
- Blosse, A.; Levy, M.; Robe, C.; Staedel, C.; Copie-Bergman, C.; Lehours, P. Deregulation of miRNA in Helicobacter pylori-induced gastric MALT lymphoma: From mice to human. J. Clin. Med. 2019, 8, 845. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef]
- Koch, M.; Mollenkopf, H.-J.; Klemm, U.; Meyer, T.F. Induction of microRNA-155 Is TLR-and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc. Natl. Acad. Sci. USA 2012, 109, E1153–E1162. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xiao, B.; Liu, Z.; Li, N.; Zhu, E.-D.; Li, B.-S.; Xie, Q.-H.; Zhuang, Y.; Zou, Q.-M.; Mao, X.-H. Identification of MyD88 as a novel target of MiR-155, involved in negative regulation of Helicobacter pylori-induced inflammation. FEBS Lett. 2010, 584, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 Modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Liu, Z.; Li, B.-S.; Tang, B.; Li, W.; Guo, G.; Shi, Y.; Wang, F.; Wu, Y.; Tong, W.-D.; et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J. Infect. Dis. 2009, 200, 916–925. [Google Scholar] [CrossRef]
- Oertli, M.; Engler, D.B.; Kohler, E.; Koch, M.; Meyer, T.F.; Müller, A. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic gastritis and colitis. J. Immunol. 2011, 187, 3578–3586. [Google Scholar] [CrossRef]
- Huffaker, T.B.; Lee, S.-H.; Tang, W.W.; Wallace, J.A.; Alexander, M.; Runtsch, M.C.; Larsen, D.K.; Thompson, J.; Ramstead, A.G.; Voth, W.P.; et al. Antitumor immunity is defective in T cell-specific microRNA-155-deficient mice and is rescued by immune checkpoint blockade. J. Biol. Chem. 2017, 292, 18530–18541. [Google Scholar] [CrossRef]
- Lario, S.; Ramírez-Lázaro, M.J.; Aransay, A.M.; Lozano, J.J.; Montserrat, A.; Casalots, Á.; Junquera, F.; Álvarez, J.; Segura, F.; Campo, R.; et al. MicroRNA profiling in duodenal ulcer disease caused by Helicobacter pylori infection in a western population. Clin. Microbiol. Infect. 2012, 18, E273–E282. [Google Scholar] [CrossRef][Green Version]
- Cortés-Márquez, A.C.; Mendoza-Elizalde, S.; Arenas-Huertero, F.; Trillo-Tinoco, J.; Valencia-Mayoral, P.; Consuelo-Sánchez, A.; Zarate-Franco, J.; Dionicio-Avendaño, A.R.; Herrera-Esquivel, J.; de Recinos-Carrera, J.E.G.; et al. Differential expression of miRNA-146a and miRNA-155 in gastritis induced by Helicobacter pylori infection in paediatric patients, adults, and an animal model. BMC Infect. Dis. 2018, 18, 463. [Google Scholar] [CrossRef]
- Krishnan, J.; Selvarajoo, K.; Tsuchiya, M.; Lee, G.; Choi, S. Toll-like receptor signal transduction. Exp. Mol. Med. 2007, 39, 421–438. [Google Scholar] [CrossRef]
- Nahid, M.A.; Satoh, M.; Chan, E.K.L. Mechanistic Role of microRNA-146a in Endotoxin-induced differential cross-regulation of TLR signaling. J. Immunol. 2011, 186, 1723–1734. [Google Scholar] [CrossRef]
- Nahid, M.A.; Yao, B.; Dominguez-Gutierrez, P.R.; Kesavalu, L.; Satoh, M.; Chan, E.K.L. Regulation of TLR2-mediated tolerance and cross-tolerance through IRAK4 modulation by MiR-132 and MiR-212. J. Immunol. 2013, 190, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, B.; Tang, B.; Li, B.; Li, N.; Zhu, E.; Guo, G.; Gu, J.; Zhuang, Y.; Liu, X.; et al. Up-regulated microRNA-146a negatively modulate Helicobacter pylori-induced inflammatory response in human gastric epithelial cells. Microbes Infect. 2010, 12, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.P.; Pereira, J.N.; De Labio, R.W.; Carneiro, L.C.; Pontes, J.C.; Barbosa, M.S.; Smith, M.D.A.C.; Payão, S.L.M.; Rasmussen, L.T. Decrease of MiR-125a-5p in gastritis and gastric cancer and its possible association with H. pylori. J. Gastrointest. Cancer 2020. [Google Scholar] [CrossRef]
- Cao, Y.; Tan, S.; Tu, Y.; Zhang, G.; Liu, Y.; Li, D.; Xu, S.; Le, Z.; Xiong, J.; Zou, W.; et al. MicroRNA-125a-5p Inhibits invasion and metastasis of gastric cancer cells by targeting BRMS1 expression. Oncol. Lett. 2018, 15, 5119–5130. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.K.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (MiR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Zhang, Z.; DuBois, R.N. Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor. Oncogene 2001, 20, 4450–4456. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Z.; Gao, C.; Chen, P.; Chen, J.; Liu, W.; Xiao, S.; Lu, H. MiR-21 Plays a pivotal role in gastric cancer pathogenesis and progression. Lab. Investig. 2008, 88, 1358–1366. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.-A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.J.; Masters, S.L. Cutting edge: MiR-223 and EBV MiR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Palermo, R.; Talora, C.; Campese, A.F.; Checquolo, S.; Bellavia, D.; Tottone, L.; Testa, G.; Miele, E.; Indraccolo, S.; et al. Notch and NF-KB signaling pathways regulate MiR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, J.; Cheng, Y.; Jiang, Y.; Li, G. Over-Expression of MicroRNA-223 Inhibited the proinflammatory responses in Helicobacter pylori-infection macrophages by down-regulating IRAK-1. Am. J. Transl. Res. 2016, 8, 615–622. [Google Scholar] [PubMed]
- Yang, F.; Xu, Y.; Liu, C.; Ma, C.; Zou, S.; Xu, X.; Jia, J.; Liu, Z. NF-ΚB/MiR-223-3p/ARID1A axis is involved in Helicobacter pylori CagA-induced gastric carcinogenesis and progression. Cell Death Dis. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.F.T.; Cadamuro, A.C.T.; Biselli-Périco, J.M.; Leite, K.R.M.; Severino, F.E.; Reis, P.P.; Cordeiro, J.A.; Silva, A.E. Interaction between inflammatory mediators and miRNAs in Helicobacter pylori infection. Cell Microbiol. 2016, 18, 1444–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xu, J. Downregulation of MicroRNA-204 Increases the expression of matrix metallopeptidase 9 in pediatric patients with pulpitis and Helicobacter pylori infection in the stomach. Exp. Ther. Med. 2019, 18, 253–259. [Google Scholar] [CrossRef]
- Feng, J.; Guo, J.; Wang, J.-P.; Chai, B.-F. MiR-32-5p aggravates intestinal epithelial cell injury in pediatric enteritis induced by Helicobacter pylori. World J. Gastroenterol. 2019, 25, 6222–6237. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Săsăran, M.O.; Meliț, L.E.; Dobru, E.D. MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori. Int. J. Mol. Sci. 2021, 22, 1406. https://doi.org/10.3390/ijms22031406
Săsăran MO, Meliț LE, Dobru ED. MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori. International Journal of Molecular Sciences. 2021; 22(3):1406. https://doi.org/10.3390/ijms22031406
Chicago/Turabian StyleSăsăran, Maria Oana, Lorena Elena Meliț, and Ecaterina Daniela Dobru. 2021. "MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori" International Journal of Molecular Sciences 22, no. 3: 1406. https://doi.org/10.3390/ijms22031406
APA StyleSăsăran, M. O., Meliț, L. E., & Dobru, E. D. (2021). MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori. International Journal of Molecular Sciences, 22(3), 1406. https://doi.org/10.3390/ijms22031406