
Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Results
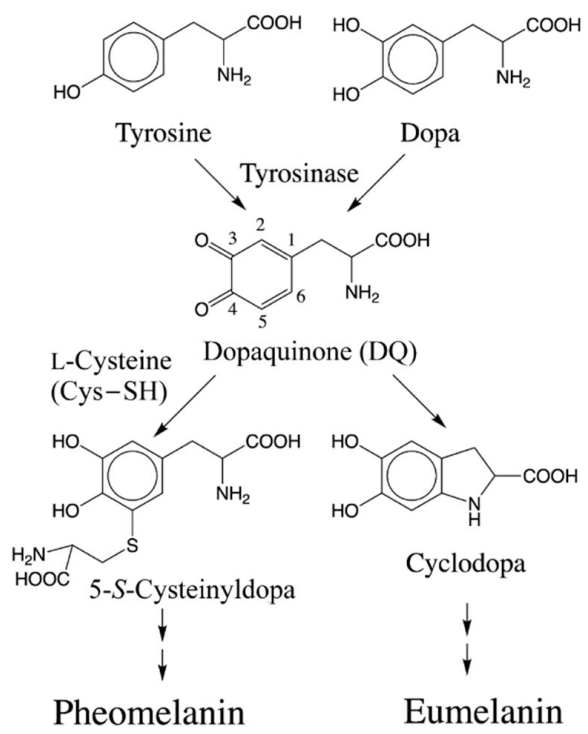
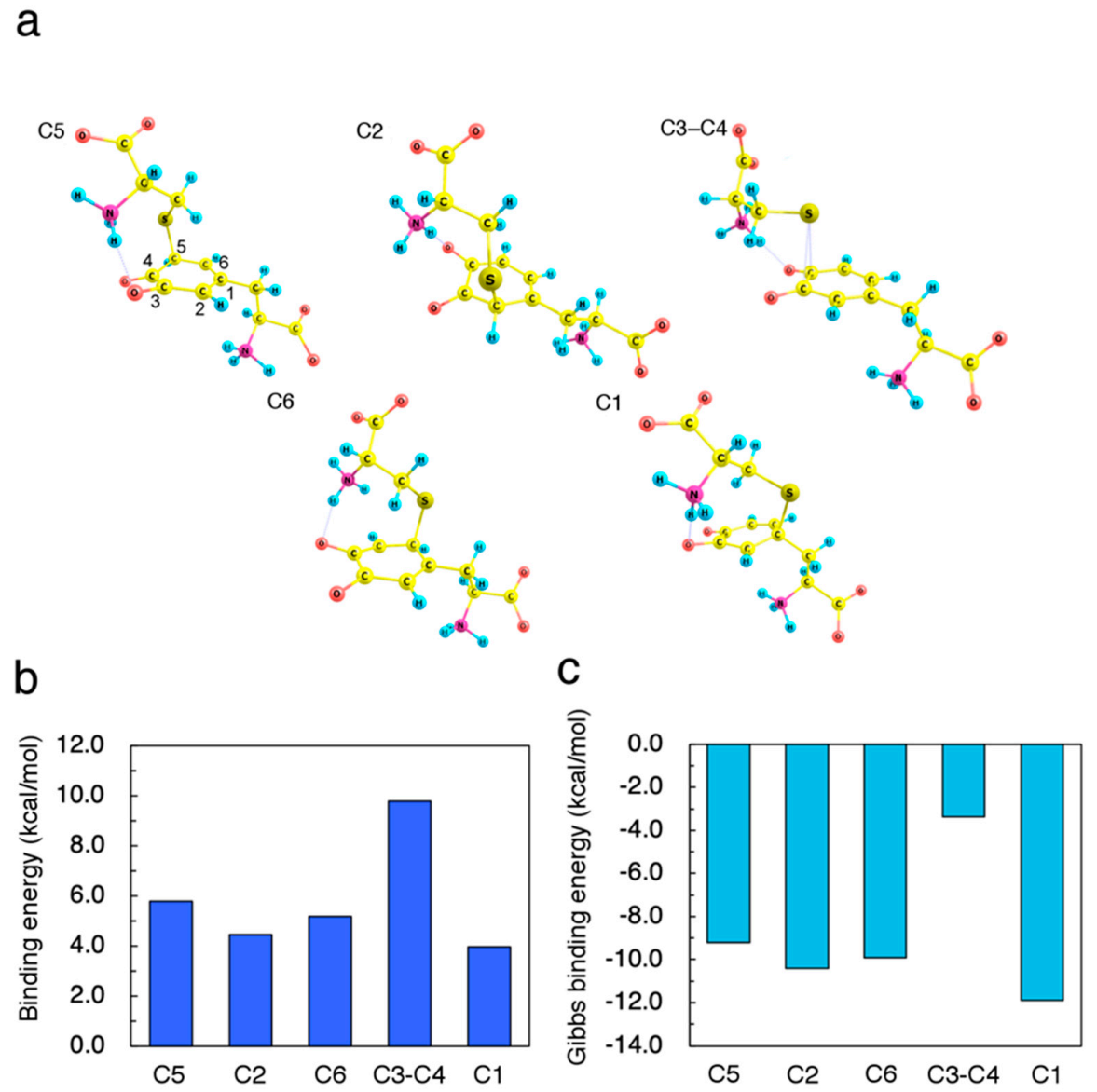
2.1. Initial Binding Sites for Cysteine Thiolate (Cys−S−) on Dopaquinone (DQ)
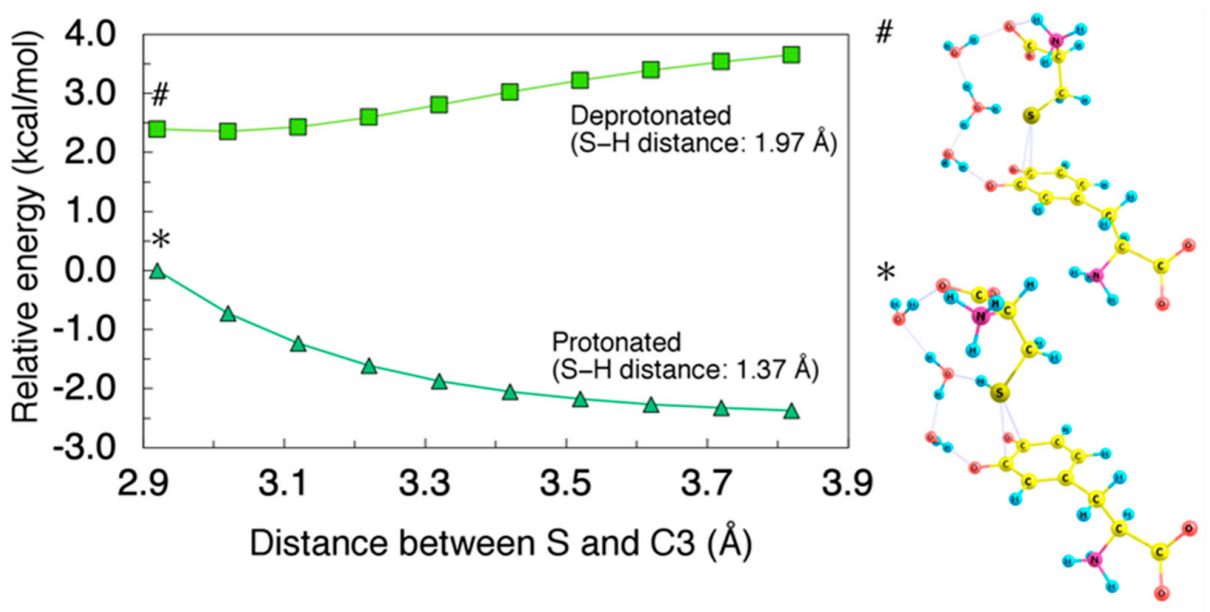
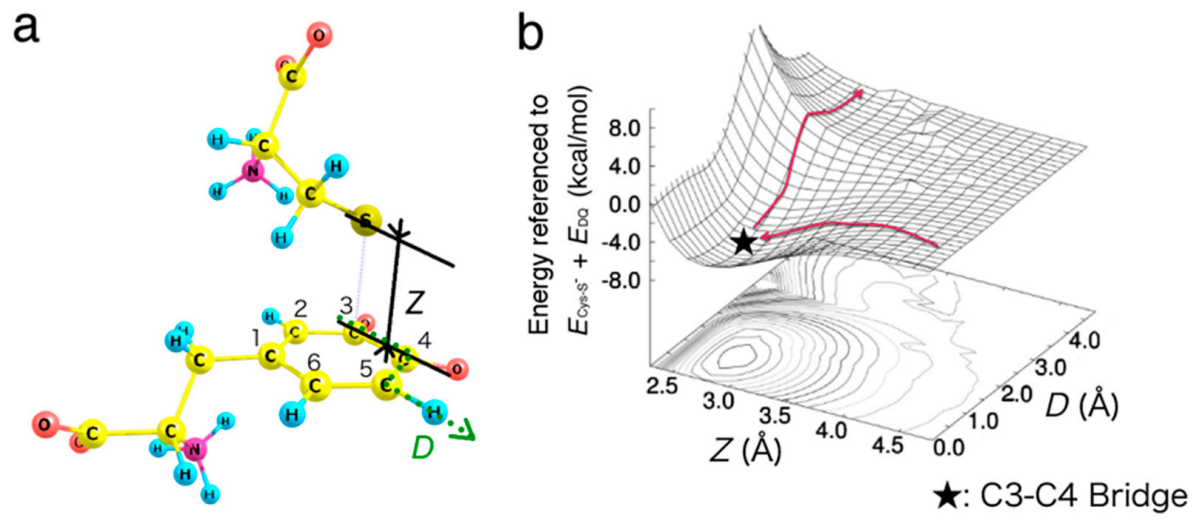
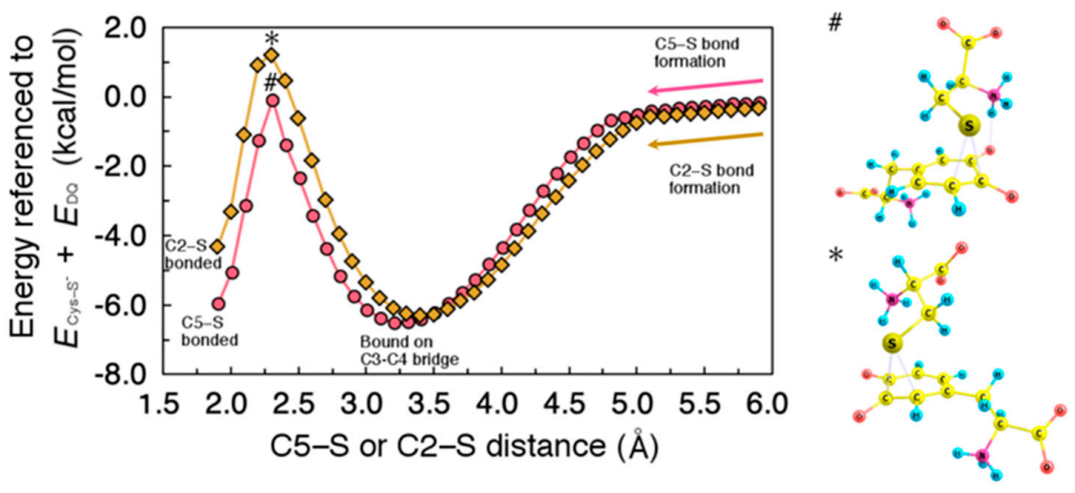
2.2. Migration of Cysteine Thiolate (Cys−S−)
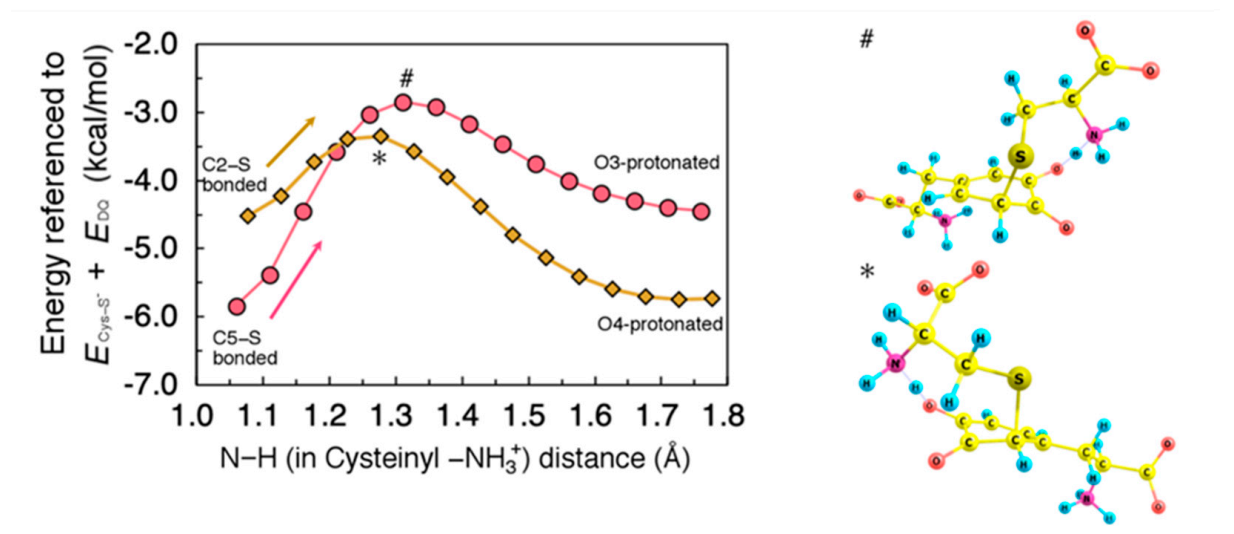
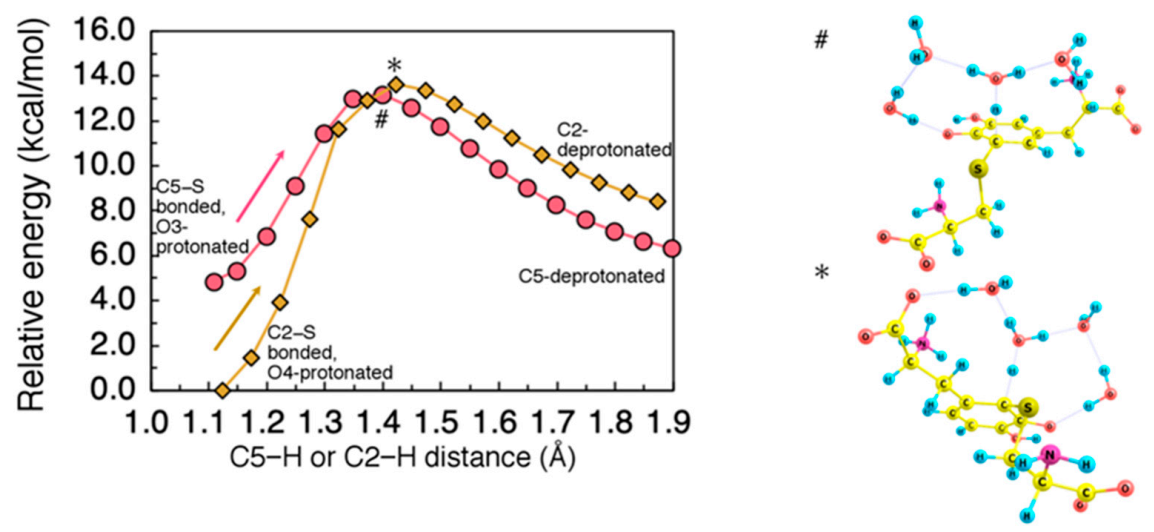
2.3. Proton Rearrangements to Give Cysteinyldopa
3. Discussion
3.1. Structural and Electronic Analyses for the Initial Intermediates
3.2. Effects of Cysteinyl Amino Group on Binding Sites and Reaction Rate
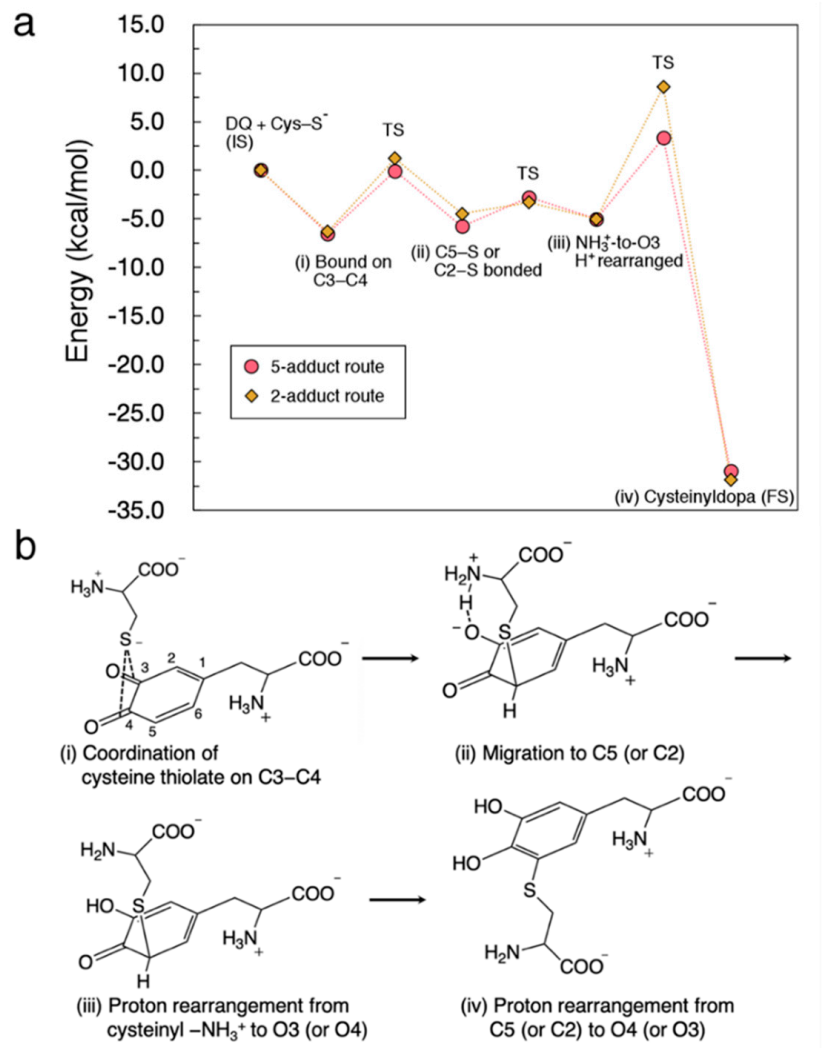
3.3. Energy Diagram for Cysteine Binding to Form Cysteinyldopa
4. Materials and Methods
4.1. Electronic State Calculation Methods
4.2. Structures for Calculations
4.3. Reaction Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cys–SH | l-cysteine |
| DFT | density functional theory |
| DQ | dopaquinone |
| HPLC | high performance liquid chromatography |
| IEF-PCM | integral equation formalism polarizable continuum model |
| LUMO | lowest unoccupied molecular orbital |
| NPA | natural population analysis |
References
- Ito, S. A chemist’s view of melanogenesis. Pigment Cell Res. 2003, 16, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis—Pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Prota, G. Recent advances in the chemistry of melanogenesis in mammals. J. Investig. Dermatol. 1980, 75, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Sugumaran, M.; Wakamatsu, K. Chemical reactivities of ortho-quinones produced in living organisms: Fate of quinonoid products formed by tyrosinase and phenoloxidase action on phenols and catechols. Int. J. Mol. Sci. 2020, 21, 6080. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803. [Google Scholar] [CrossRef]
- Sugumaran, M. Molecular mechanisms for mammalian melanogenesis. FEBS Lett. 1991, 293, 4–10. [Google Scholar] [CrossRef]
- Sugumaran, M. Reactivities of quinone methides versus o-quinones in catecholamine metabolism and eumelanin biosynthesis. Int. J. Mol. Sci. 2016, 17, 1576. [Google Scholar] [CrossRef]
- Tse, D.C.S.; McCreery, R.L.; Adams, R.N. Potential oxidative pathways of brain catecholamines. J. Med. Chem. 1976, 19, 37–40. [Google Scholar] [CrossRef]
- Cooksey, C.J.; Land, E.J.; Rushton, F.A.P.; Ramsden, C.A.; Riley, P.A. Tyrosinase-mediated cytotoxicity of 4-substituted phenols: Use of QSAR to forecast reactivities of thiols towards the derived ortho-quinones. Quant. Struct. Act. Relat. 1996, 15, 498–503. [Google Scholar] [CrossRef]
- Ito, S.; Prota, G. A facile one-step synthesis of cysteinyldopas using mushroom tyrosinase. Experientia 1977, 33, 1118–1119. [Google Scholar] [CrossRef]
- Kato, T.; Ito, S.; Fujita, K. Tyrosinase-catalyzed binding of 3,4-dihydroxyphenylalanine with proteins through the sulfhydryl group. Biochim. Biophys. Acta 1986, 881, 415–421. [Google Scholar] [CrossRef]
- Ito, S.; Kato, T.; Fujita, K. Covalent binding of catechols to proteins through the sulphydryl group. Biochem. Pharmacol. 1988, 37, 1707–1710. [Google Scholar] [CrossRef]
- Thompson, A.; Land, E.J.; Chedekel, M.R.; Subbarao, K.V.; Truscott, T.G. A pulse radiolysis investigation of the oxidation of the melanin precursors 3,4-dihydroxyphenylalanine (dopa) and the cysteinyldopas. Biochim. Biophys. Acta 1985, 843, 49–57. [Google Scholar] [CrossRef]
- Ozeki, H.; Ito, S.; Wakamatsu, K.; Hirobe, T. Chemical characterization of hair melanins in various coat-color mutants of mice. J. Investig. Dermatol. 1995, 105, 361–365. [Google Scholar] [CrossRef]
- Ozeki, H.; Ito, S.; Wakamatsu, K.; Thody, A.J. Spectrophtometric characterization of eumelanin and pheomelanin in hair. Pigment Cell Res. 1996, 9, 265–270. [Google Scholar] [CrossRef]
- Liu, Y.; Hong, L.; Wakamatsu, K.; Ito, S.; Adhyaru, B.; Cheng, C.Y.; Bowers, C.R.; Simon, J.D. Comparison of structural and chemical properties of black and red human hair melanosomes. Photochem. Photobiol. 2005, 81, 135–144. [Google Scholar] [CrossRef]
- Mårs, U.; Larsson, B.S. Pheomelanin as a binding site for drugs and chemicals. Pigment Cell Res. 1999, 12, 266–274. [Google Scholar] [CrossRef]
- Chedekel, M.R.; Post, P.W.; Deibel, R.M.; Kalus, M. Photodestruction of phaeomelanin. Photochem. Photobiol. 1977, 26, 651–653. [Google Scholar] [CrossRef]
- Chedekel, M.R.; Smith, S.K.; Post, P.W.; Pokora, A.; Vessell, D.L. Photodestruction of pheomelanin: Role of oxygen. Proc. Natl. Acad. Sci. USA 1978, 75, 5395–5399. [Google Scholar] [CrossRef]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef]
- Panzella, L.; Leone, L.; Greco, G.; Vitiello, G.; D’Errico, G.; Napolitano, A. Red human hair pheomelanin is a potent pro-oxidant mediating UV-independent contriburoty mechanisms of melanogenesis. Pigment Cell Melanoma Res. 2014, 27, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Panzella, L.; Monfrecola, G.; D’Ischia, M. Pheomelanin-induced oxidative stress: Bright and dark chemistry bridging red hair phenotype and melanoma. Pigment Cell Melanoma Res. 2014, 27, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.J.; Miserez, A.; Waite, H.J. Diverse strategies of protein sclerotization in marine invertebrates: Structure-property relationships in natural biomaterials. Adv. Insect Physiol. 2010, 38, 75–132. [Google Scholar]
- Sugumaran, M. Chemistry of cuticular sclerotization. Adv. Insect Physiol. 2010, 39, 151–209. [Google Scholar]
- Abebe, A.; Zheng, D.; Evans, J.; Sugumaran, M. Novel post-translational oligomerization of peptidyl dehydrodopa model compound, 1,2-dehydro-N-acetyldopa methyl ester. Bioorg. Chem. 2016, 66, 33–40. [Google Scholar] [CrossRef]
- Sugumaran, M.; Robinson, W. Structure, biosynthesis and possible function of tunichromes and related compounds. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 1–25. [Google Scholar] [CrossRef]
- Roman, T.; Diño, W.A.; Nakanishi, H.; Kasai, H. Amino acid adsorption effects on nanotube electronics. J. Vac. Soc. Jpn. 2006, 49, 440–442. [Google Scholar] [CrossRef]
- Roman, T.; Diño, W.A.; Nakanishi, H.; Kasai, H. Amino acid adsorption on single-walled carbon nanotubes. Eur. Phys. J. D 2006, 38, 117–120. [Google Scholar] [CrossRef]
- Kishida, R.; Ushijima, Y.; Saputro, A.G.; Kasai, H. Effect of pH on elementary steps of dopachrome conversion from first-principles calculation. Pigment Cell Melanoma Res. 2014, 27, 734–743. [Google Scholar] [CrossRef]
- Kishida, R.; Saputro, A.G.; Kasai, H. Mechanism of dopachrome tautomerization into 5,6-dihydroxyindole-2-carboxylic acid catalyzed by Cu(II) based on quantum chemical calculations. Biochim. Biophys. Acta 2015, 1850, 281–286. [Google Scholar] [CrossRef]
- Kishida, R.; Kasai, H.; Aspera, S.M.; Arevalo, R.L.; Nakanishi, H. Branching reaction in melanogenesis: The effect of intramolecular cyclization on thiol binding. J. Electron. Mater. 2017, 46, 3784–3788. [Google Scholar] [CrossRef]
- Kishida, R.; Kasai, H.; Aspera, S.M.; Arevalo, R.L.; Nakanishi, H. Density functional theory-based first principles calculations of rhododendrol-quinone reactions: Preference to thiol binding over cyclization. J. Phys. Soc. Jpn. 2017, 86, 024804. [Google Scholar] [CrossRef]
- Kishida, R.; Saputro, A.G.; Arevalo, R.L.; Kasai, H. Effects of introduction of α-carboxylate, N-methyl, and N-formyl groups on intramolecular cyclization of o-quinone amines: Density functional theory-based study. Int. J. Quant. Chem. 2017, 117, e25445. [Google Scholar] [CrossRef]
- Kishida, R.; Kasai, H. Cyclic bond formation of rhododendrol-quinone and dopamine-quinone: Effects of proton rearrangement. J. Phys. Soc. Jpn. 2018, 87, 084802. [Google Scholar] [CrossRef]
- Kishida, R.; Kasai, H. Computational Materials Design Case Study III: Biosynthesis of Melanin Pigment; Osaka University Press: Osaka, Japan, 2019. (In Japanese) [Google Scholar]
- Kishida, R.; Aspera, S.M.; Kasai, H. Melanin chemistry explored by quantum mechanics. Springer-Nature. (in preparation).
- Huang, X.; Xu, R.; Hawley, M.D.; Hopkins, T.L.; Kramer, K.J. Electrochemical oxidation of N-acyldopamines and regioselective reactions of their quinones with N-acetylcysteine and thiourea. Arch. Biochem. Biophys. 1988, 352, 19–30. [Google Scholar] [CrossRef][Green Version]
- Xu, R.; Huang, X.; Kramer, K.J.; Hawley, M.D. Characterization of products from the reactions of dopamine quinone with N-acetylcysteine. Bioorg. Chem. 1996, 24, 110–126. [Google Scholar] [CrossRef]
- Jameson, G.N.L.; Zhang, J.; Jameson, R.F.; Linert, W. Kinetic evidence that cysteine reacts with dopaminoquinone via reversible adduct formation to yield 5-cysteinyl-dopamine: An important precursor of neuromelanin. Org. Biomol. Chem. 2004, 2, 777–782. [Google Scholar] [CrossRef]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. ortho-Quinone amines and derivatives: The influence of structure on the rates and modes of intramolecular reaction. Arkivoc 2007, xi, 23–36. [Google Scholar]
- Becke, A.D. Density-functional thermocheimstry. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Sugumaran, M.; Dali, H.; Semensi, V. Chemical- and cuticular phenoloxidase-mediated synthesis of cysteinyl-catechol adducts. Arch. Insect Biochem. Physiol. 1989, 11, 127–137. [Google Scholar] [CrossRef]
- Ito, S.; Palumbo, A.; Prota, G. Tyrosinase-catalyzed conjugation of dopa with glutathione. Experientia 1985, 41, 960–961. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ohtara, K.; Ito, S. Chemical analysis of late stages of pheomelanogenesis: Conversion of dihydrobenzothiazine to a benzothiazole structure. Pigment Cell Melanoma Res. 2009, 22, 474–486. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Revision C. 01); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Site a | C−S Bond Length (Å) b | O3-C3-C4-O4 Dihedral Angle (Deg.) c |
|---|---|---|
| Before reaction | N/A | 0.27 |
| C5 | 1.91 | 25.59 |
| C2 | 1.92 | 25.19 |
| C6 | 1.95 | 10.44 |
| C3−C4 | 2.75 | 4.05 |
| C1 | 1.96 | 13.33 |
| Binding Site | Natural Charges (e) | ||
|---|---|---|---|
| S | O3 | O4 | |
| Before reaction | −0.73 | −0.54 | −0.53 |
| C5 | 0.19 | −0.80 | −0.61 |
| C2 | 0.19 | −0.63 | −0.81 |
| C6 | 0.15 | −0.63 | −0.83 |
| C3–C4 | −0.28 | −0.65 | −0.70 |
| C1 | 0.15 | −0.82 | −0.62 |
| Binding Site | Reaction a | Relative Energy (kcal/mol) b |
|---|---|---|
| C5 | Cys−SH + DQ → Cys−S· + DQ−H· (at O3) | 12.9 |
| Cys−S· + DQ−H· → Cys−S·/DQ−H· | 20.9 | |
| Cys−S·/DQ−H· → Cys−S/DQ−H | −5.0 | |
| C3−C4 | Cys−SH + DQ → Cys−S· + DQ−H· (at O3) | 12.9 |
| Cys−S· + DQ−H· → Cys−S·/DQ−H· | Not bonded c | |
| Cys−S·/DQ−H· → Cys−S/DQ−H | −9.2 | |
| C6 | Cys−SH + DQ → Cys−S· + DQ−H· (at O4) | 13.4 |
| Cys−S· + DQ−H· → Cys−S·/DQ−H· | Not bonded c | |
| Cys−S·/DQ−H· → Cys−S/DQ−H | −12.9 |
| Binding Site | ΔEb = Eb, Not HB − Eb, HB (kcal/mol) a |
|---|---|
| C5 | 2.8 |
| C2 | 4.6 |
| C6 | 1.0 |
| C3–C4 | 3.4 |
| C1 | 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishida, R.; Ito, S.; Sugumaran, M.; Arevalo, R.L.; Nakanishi, H.; Kasai, H. Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone. Int. J. Mol. Sci. 2021, 22, 1373. https://doi.org/10.3390/ijms22031373
Kishida R, Ito S, Sugumaran M, Arevalo RL, Nakanishi H, Kasai H. Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone. International Journal of Molecular Sciences. 2021; 22(3):1373. https://doi.org/10.3390/ijms22031373
Chicago/Turabian StyleKishida, Ryo, Shosuke Ito, Manickam Sugumaran, Ryan Lacdao Arevalo, Hiroshi Nakanishi, and Hideaki Kasai. 2021. "Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone" International Journal of Molecular Sciences 22, no. 3: 1373. https://doi.org/10.3390/ijms22031373
APA StyleKishida, R., Ito, S., Sugumaran, M., Arevalo, R. L., Nakanishi, H., & Kasai, H. (2021). Density Functional Theory-Based Calculation Shed New Light on the Bizarre Addition of Cysteine Thiol to Dopaquinone. International Journal of Molecular Sciences, 22(3), 1373. https://doi.org/10.3390/ijms22031373