
A DNA Sequence Based Polymer Model for Chromatin Folding

Abstract
1. Introduction
2. Results
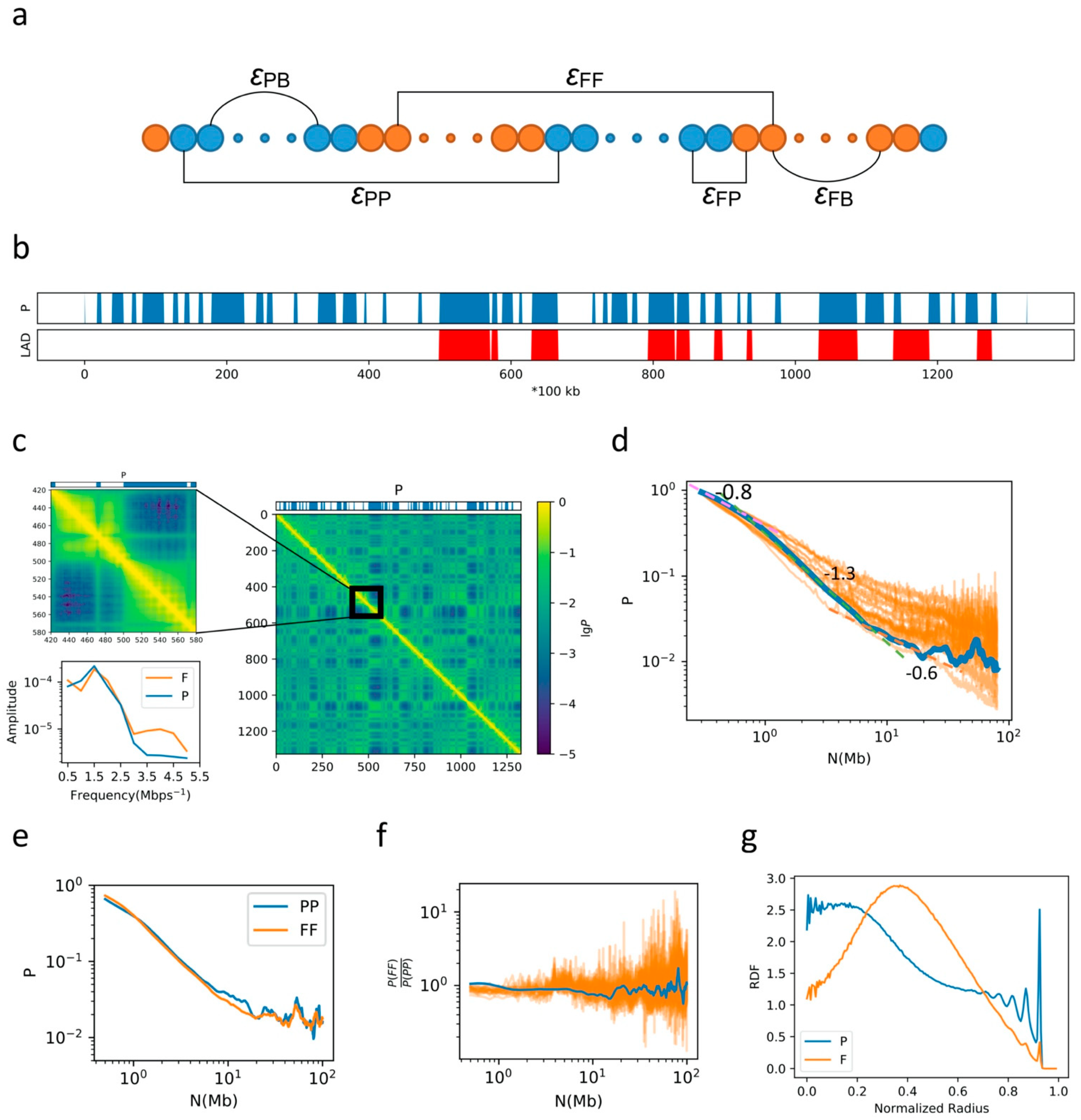
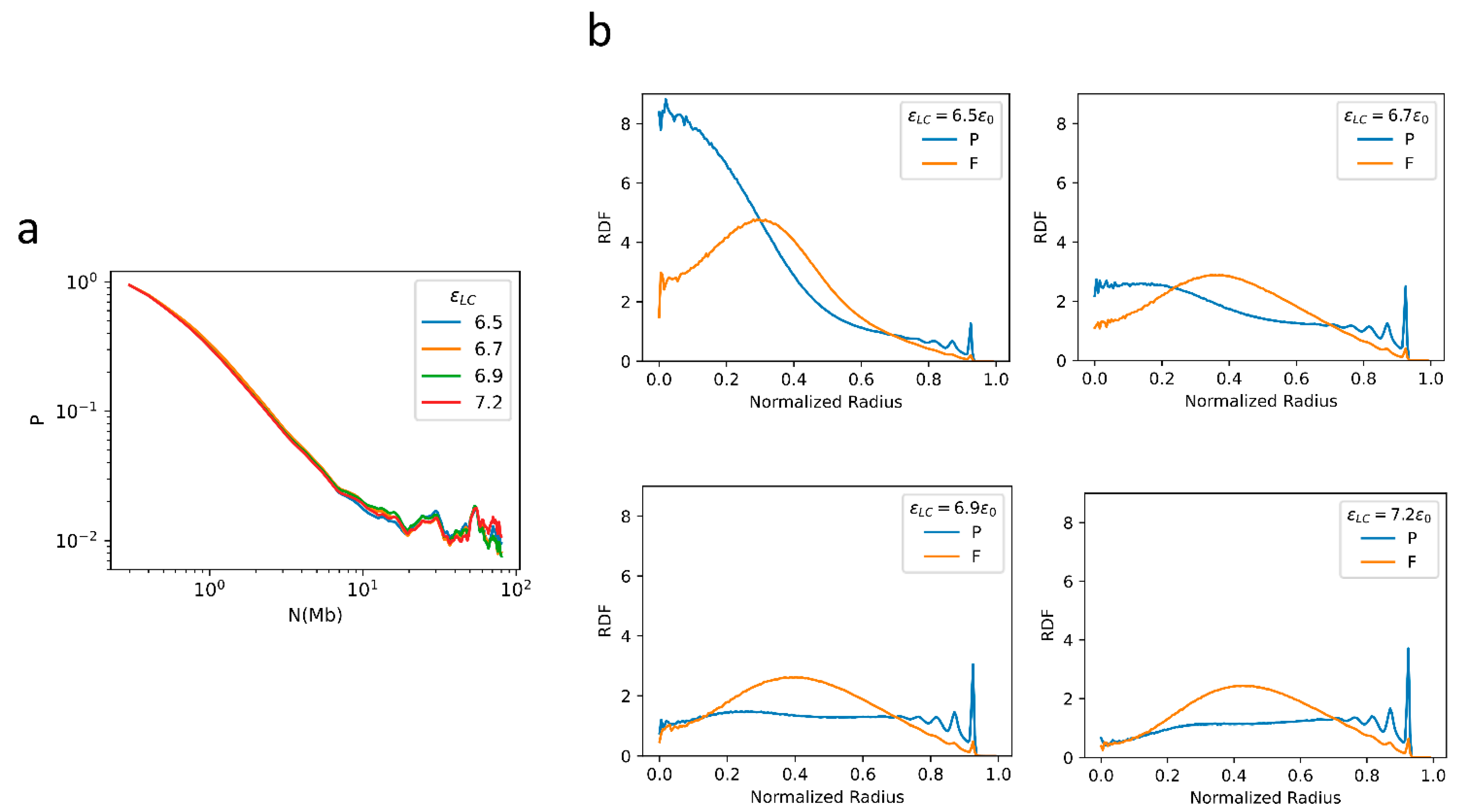
2.1. General Properties of Chromatin Folding Are Reproduced by Our Model
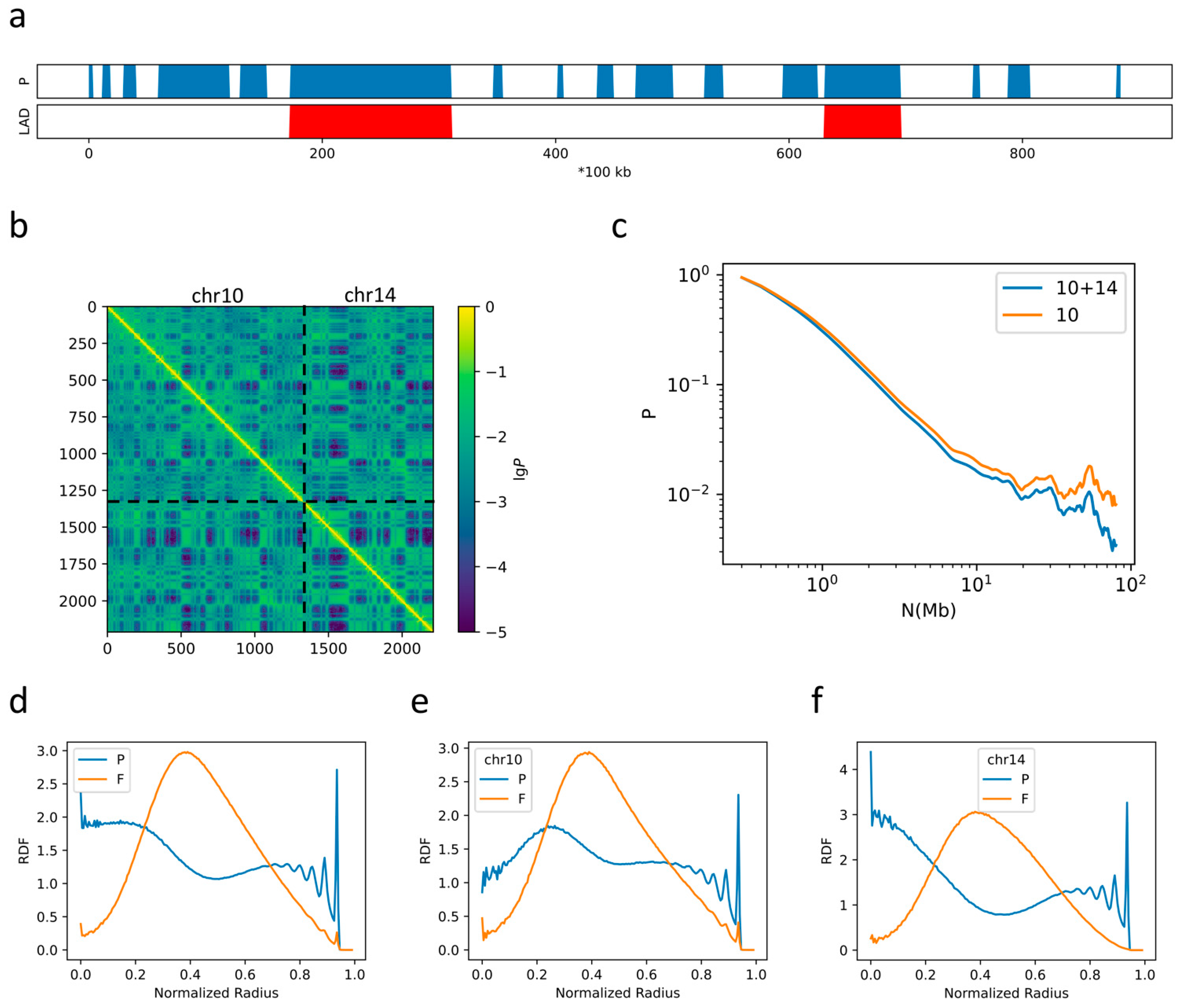
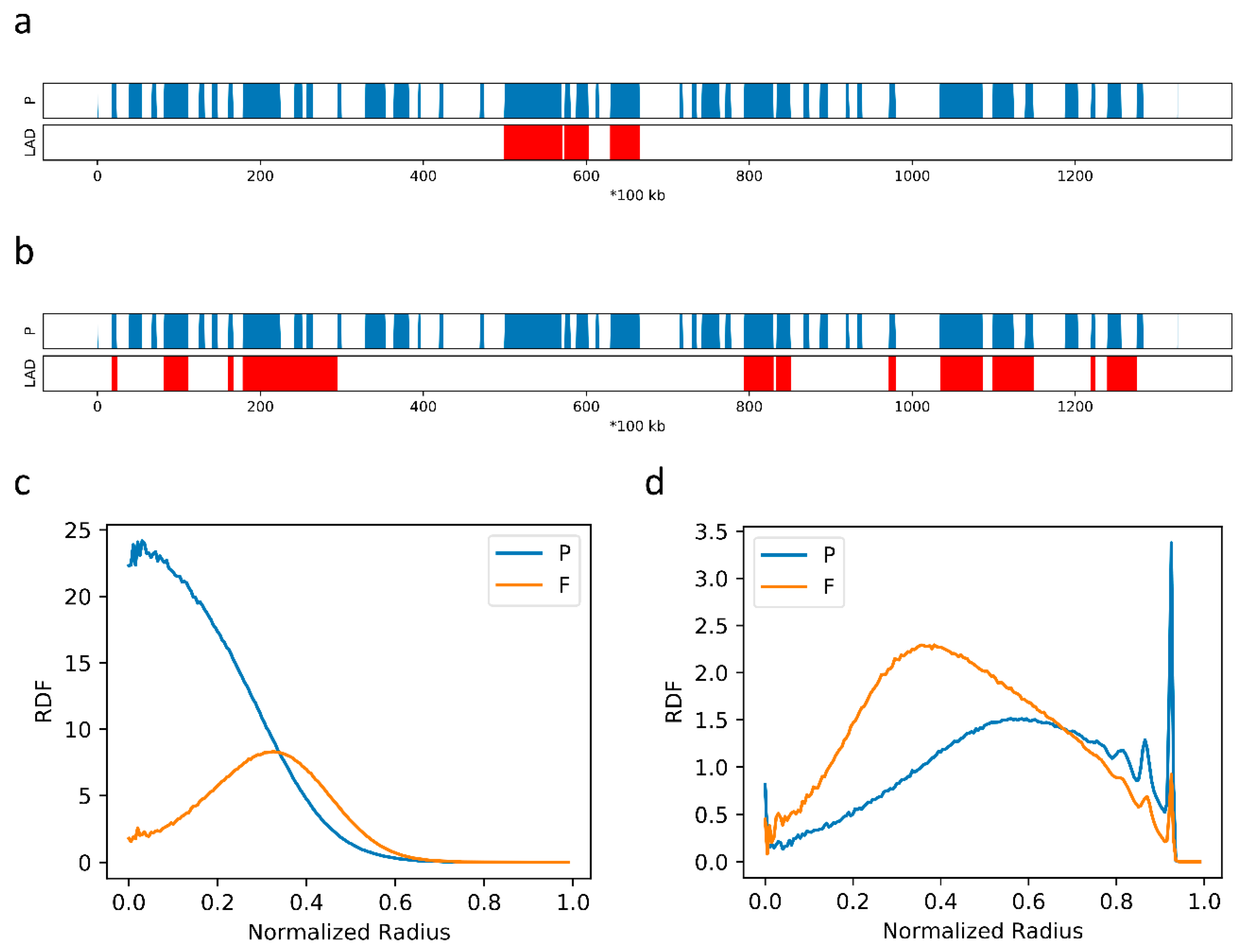
2.2. The Formation of Chromosome Territories Is Partially Reproduced
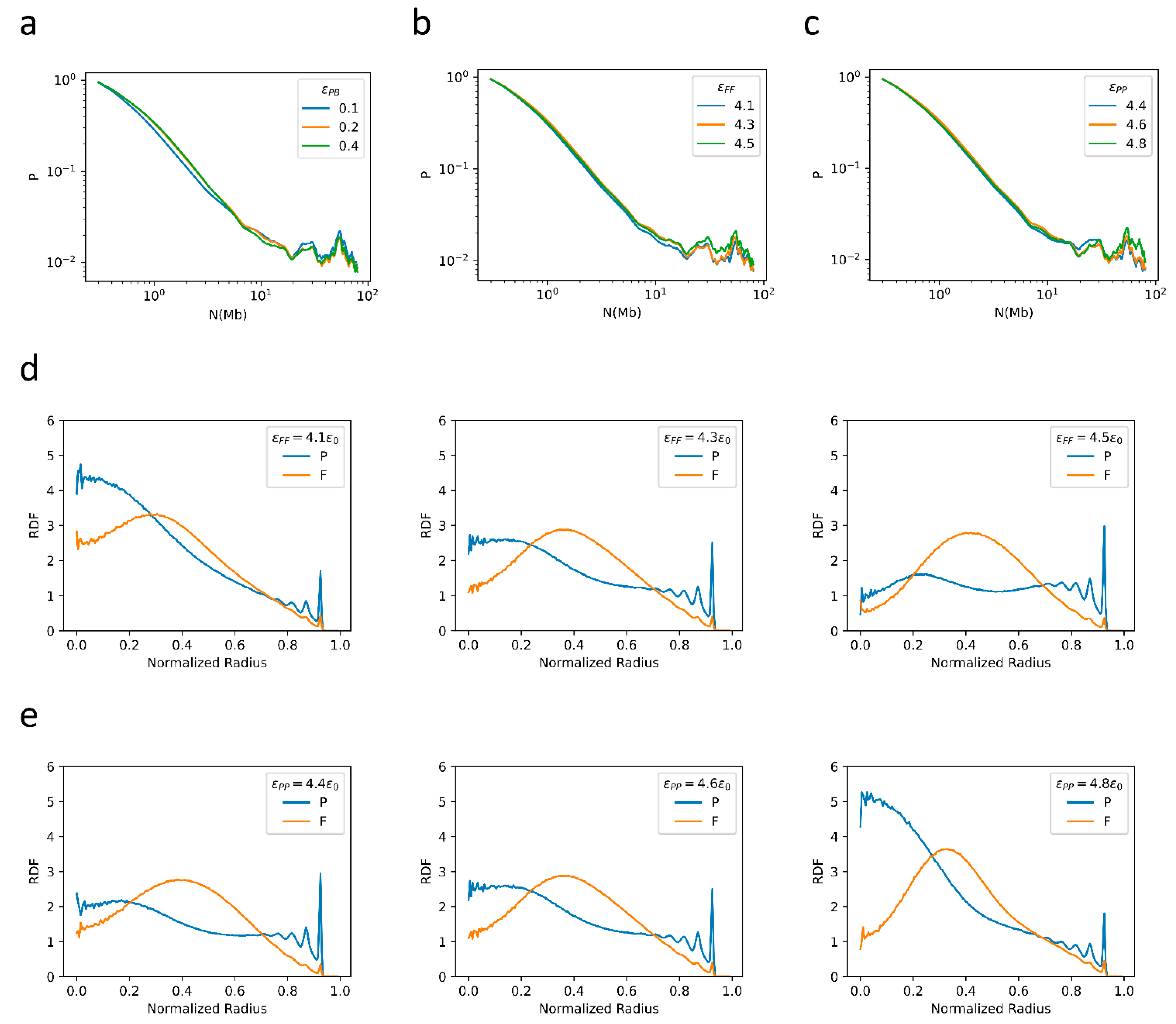
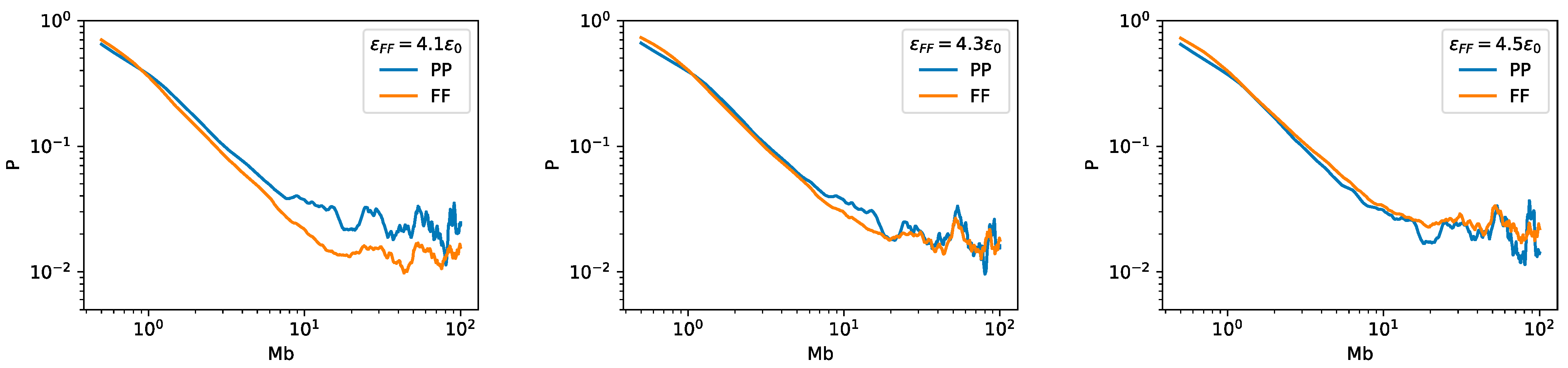
2.3. Bonding between TAD Boundaries Affects the Decay of Contact Probability with Genomic Distance
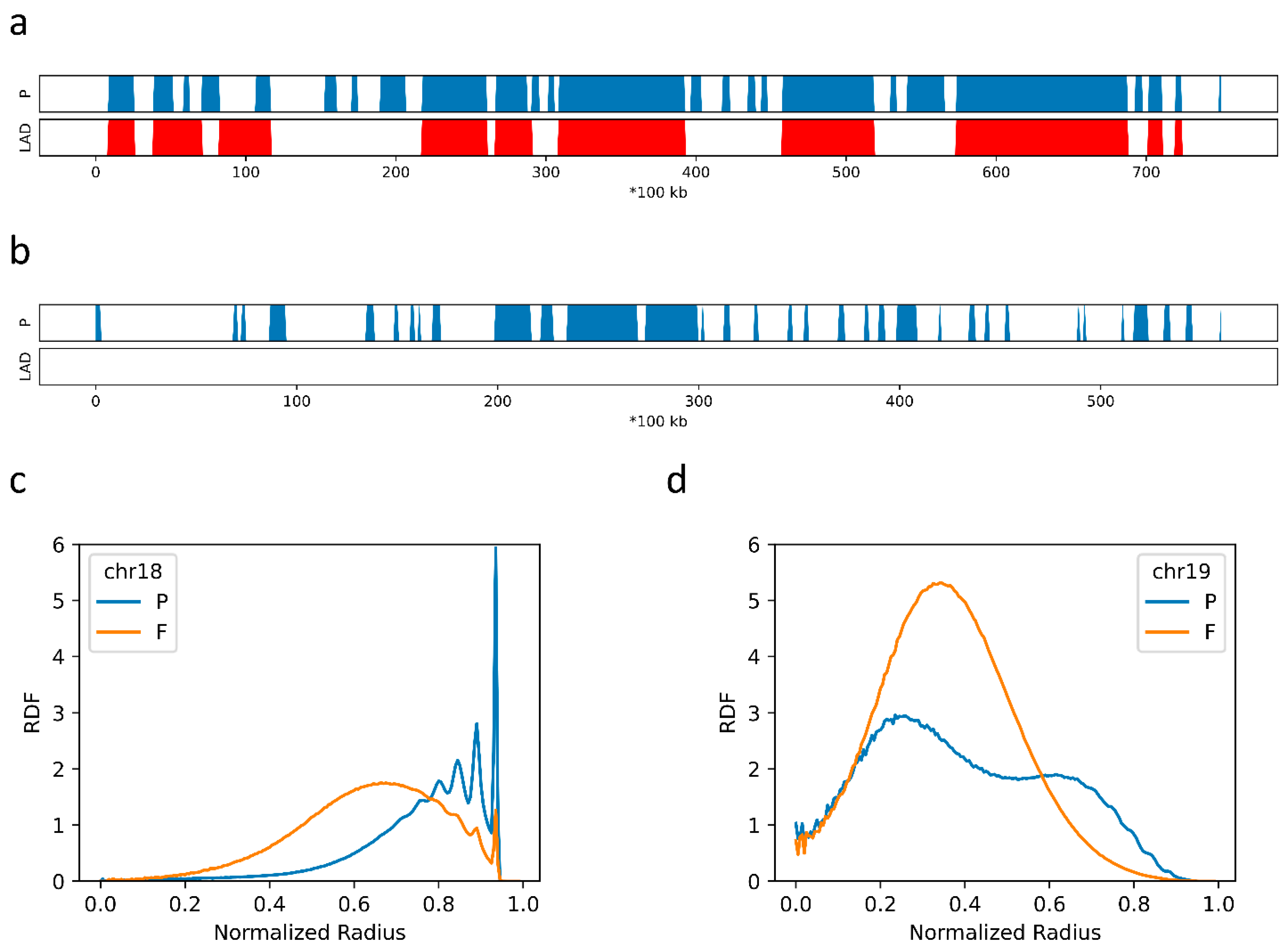
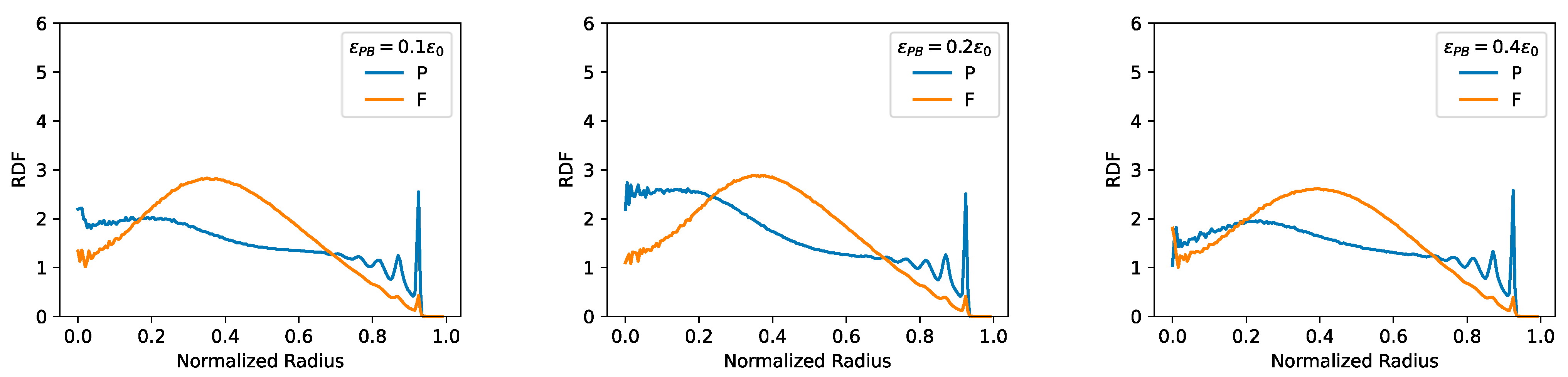
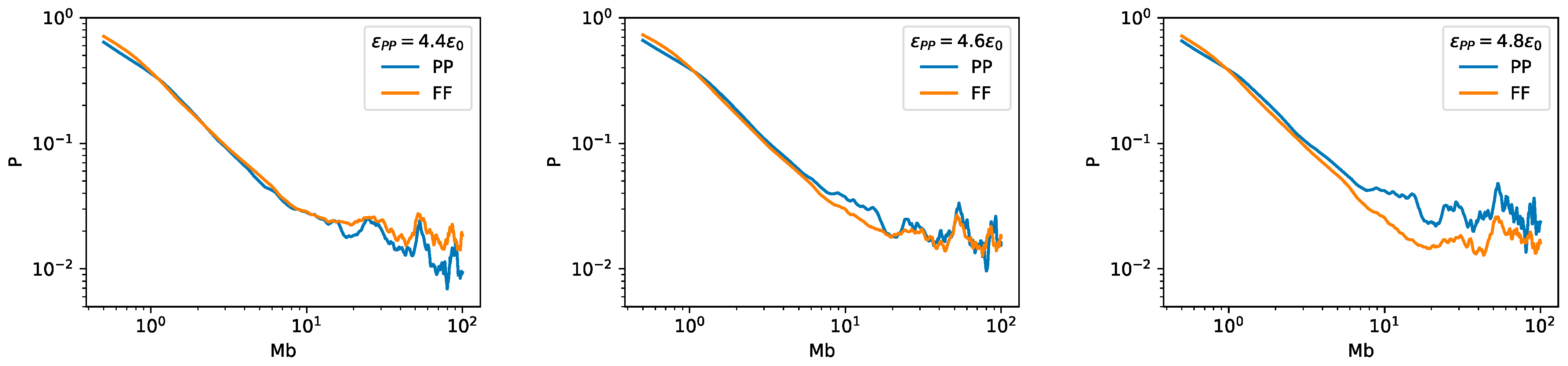
2.4. F–F, P–P Interactions and LAD Formation Mainly Affect Spatial Organization of Chromatin
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Xie, W.J.; Meng, L.; Liu, S.; Zhang, L.; Cai, X.; Gao, Y.Q. Structural Modeling of Chromatin Integrates Genome Features and Reveals Chromosome Folding Principle. Sci. Rep. 2017, 7, 2818. [Google Scholar] [CrossRef]
- Jin, F.; Li, Y.; Dixon, J.R.; Selvaraj, S.; Ye, Z.; Lee, A.Y.; Yen, C.A.; Schmitt, A.D.; Espinoza, C.A.; Ren, B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013, 503, 290–294. [Google Scholar] [CrossRef]
- Finn, E.H.; Pegoraro, G.; Brandão, H.B.; Valton, A.L.; Oomen, M.E.; Dekker, J.; Mirny, L.; Misteli, T. Extensive Heterogeneity and Intrinsic Variation in Spatial Genome Organization. Cell 2019, 176, 1502–1515.e10. [Google Scholar] [CrossRef]
- Sanborn, A.L.; Rao, S.S.P.; Huang, S.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. Chromosome Architecture and Genome Organization. PLoS ONE 2015, 10, 0143739. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The formation of chromatin domains: A new model. bioRxiv 2018. [Google Scholar] [CrossRef]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.H.; Haering, C.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Luperchio, T.; Sauria, M.; Hoskins, V.; Wong, X.; DeBoy, E.; Gaillard, M.C.; Tsang, P.; Pekrun, K.; Ach, R.; Yamada, N.; et al. The repressive genome compartment is established early in the cell cycle before forming the lamina associated domains. bioRxiv 2018. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, L.; Quan, H.; Tian, H.; Meng, L.; Yang, L.; Feng, H.; Gao, Y.Q. From 1D sequence to 3D chromatin dynamics and cellular functions: A phase separation perspective. Nucleic Acids Res. 2018, 46, 9367–9383. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-Cell Dynamics of Genome-Nuclear Lamina Interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-wide Maps of Nuclear Lamina Interactions in Single Human Cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling epigenome folding: Formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014, 42, 9553–9561. [Google Scholar] [CrossRef] [PubMed]
- Brackley, C.A.; Brown, J.M.; Waithe, D.; Babbs, C.; Davies, J.; Hughes, J.R.; Buckle, V.J.; Marenduzzo, D. Predicting the three-dimensional folding of cis-regulatory regions in mammalian genomes using bioinformatic data and polymer models. Genome Biol. 2016, 17, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Michieletto, D.; Orlandini, E.; Marenduzzo, D. Polymer model with Epigenetic Recoloring Reveals a Pathway for the de novo Establishment and 3D Organization of Chromatin Domains. Phys. Rev. X 2016, 6, 041047. [Google Scholar] [CrossRef]
- Shi, G.; Liu, L.; Hyeon, C.; Thirumalai, D. Interphase human chromosome exhibits out of equilibrium glassy dynamics. Nat. Commun. 2018, 9, 3161. [Google Scholar] [CrossRef]
- Barbieri, M.; Chotalia, M.; Fraser, J.; Lavitas, L.M.; Dostie, J.; Pombo, A.; Nicodemi, M. Complexity of chromatin folding is captured by the strings and binders switch model. Proc. Natl. Acad. Sci. USA 2012, 109, 16173–16178. [Google Scholar] [CrossRef]
- Brackley, C.A.; Taylor, S.; Papantonis, A.; Cook, P.R.; Marenduzzo, D. Nonspecific bridging-induced attraction drives clustering of DNA-binding proteins and genome organization. Proc. Natl. Acad. Sci. USA 2013, 110, E3605–E3611. [Google Scholar] [CrossRef]
- Brackley, C.A.; Johnson, J.; Kelly, S.; Cook, P.R.; Marenduzzo, D. Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucleic Acids Res. 2016, 44, 3503–3512. [Google Scholar] [CrossRef]
- Michieletto, D.; Chiang, M.; Coli, D.; Papantonis, A.; Orlandini, E.; Cook, P.R.; Marenduzzo, D. Shaping epigenetic memory via genomic bookmarking. Nucleic Acids Res. 2018, 46, 83–93. [Google Scholar] [CrossRef]
- Chiariello, A.M.; Annunziatella, C.; Bianco, S.; Esposito, A.; Nicodemi, M. Polymer physics of chromosome large-scale 3D organisation. Sci. Rep. 2016, 6, 29775. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Reports 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.; Michieletto, D.; Brackley, C.A.; Rattanavirotkul, N.; Mohammed, H.; Marenduzzo, D.; Chandra, T. Polymer Modeling Predicts Chromosome Reorganization in Senescence. Cell Rep. 2019, 18, 3212–3223.e6. [Google Scholar] [CrossRef] [PubMed]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538.e9. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef]
- Dixon, J.R.; Xu, J.; Dileep, V.; Zhan, Y.; Song, F.; Le, V.T.; Yardımcı, G.G.; Chakraborty, A.; Bann, D.V.; Wang, Y.; et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018, 50, 1388–1398. [Google Scholar] [CrossRef]
- Huang, K.; Li, Y.; Shim, A.R.; Virk, R.K.A.; Agrawal, V.; Eshein, A.; Nap, R.J.; Almassalha, L.M.; Backman, V.; Szleifer, I. Physical and data structure of 3D genome. Sci. Adv. 2020, 6, eaay4055. [Google Scholar] [CrossRef]
- Bohn, M.; Heermann, D.W. Diffusion-Driven Looping Provides a Consistent Framework for Chromatin Organization. PLoS ONE 2010, 5, e12218. [Google Scholar] [CrossRef]
- Olarte-Plata, J.D.; Haddad, N.; Vaillant, C.; Jost, D. The folding landscape of the epigenome. Phys. Biol. 2016, 13, 026001. [Google Scholar] [CrossRef]
- Park, S.H.; Park, S.H.; Kook, M.C.; Kim, E.Y.; Park, S.; Lim, J.H. Ultrastructure of Human Embryonic Stem Cells and Spontaneous and Retinoic Acid-Induced Differentiating Cells. Ultrastruct. Pathol. 2004, 28, 229–238. [Google Scholar] [CrossRef]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic Plasticity of Chromatin Proteins in Pluripotent Embryonic Stem Cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global Transcription in Pluripotent Embryonic Stem Cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct Epigenomic Landscapes of Pluripotent and Lineage-Committed Human Cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1αsuggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef]
- Malhas, A.; Lee, C.F.; Sanders, R.; Saunders, N.J.; Vaux, D.J. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J. Cell Biol. 2007, 176, 593–603. [Google Scholar] [CrossRef]
- Camps, J.; Wangsa, D.; Falke, M.; Brown, M.; Case, C.M.; Erdos, M.R.; Ried, T. Loss of lamin B1 results in prolongation of S phase and decondensation of chromosome territories. FASEB J. 2014, 28, 3423–3434. [Google Scholar] [CrossRef]
- Chang, L.; Li, M.; Shao, S.; Li, C.; Ai, S.; Xue, B.; Hou, Y.; Zhang, Y.; Li, R.; Fan, X.; et al. Nuclear peripheral chromatin-lamin B1 interaction is required for global integrity of chromatin architecture and dynamics in human cells. Protein Cell 2020. [Google Scholar] [CrossRef]
- Gilbert, N.; Gilchrist, S.; Bickmore, W.A. Chromatin Organization in the Mammalian Nucleus. Int. Rev. Cytol. 2004, 262, 283–336. [Google Scholar]
- Rosa, A.; Everaers, R. Structure and Dynamics of Interphase Chromosomes. PLoS Comput. Biol. 2008, 4, e1000153. [Google Scholar] [CrossRef]
- Brackley, C.A.; Liebchen, B.; Michieletto, D.; Mouvet, F.; Cook, P.R.; Marenduzzo, D. Ephemeral Protein Binding to DNA Shapes Stable Nuclear Bodies and Chromatin Domains. Biophys. J. 2017, 112, 1085–1093. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Liu, C.; Cheng, Y.J.; Wang, J.W.; Weigel, D. Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat. Plants 2017, 3, 742–748. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.; Gao, Y.Q. A DNA Sequence Based Polymer Model for Chromatin Folding. Int. J. Mol. Sci. 2021, 22, 1328. https://doi.org/10.3390/ijms22031328
Zhou R, Gao YQ. A DNA Sequence Based Polymer Model for Chromatin Folding. International Journal of Molecular Sciences. 2021; 22(3):1328. https://doi.org/10.3390/ijms22031328
Chicago/Turabian StyleZhou, Rui, and Yi Qin Gao. 2021. "A DNA Sequence Based Polymer Model for Chromatin Folding" International Journal of Molecular Sciences 22, no. 3: 1328. https://doi.org/10.3390/ijms22031328
APA StyleZhou, R., & Gao, Y. Q. (2021). A DNA Sequence Based Polymer Model for Chromatin Folding. International Journal of Molecular Sciences, 22(3), 1328. https://doi.org/10.3390/ijms22031328