
Identification of New Genes Involved in Germline Predisposition to Early-Onset Gastric Cancer

 , , , , , ,
, , , , , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Clinico-Pathological Features of the Cohort
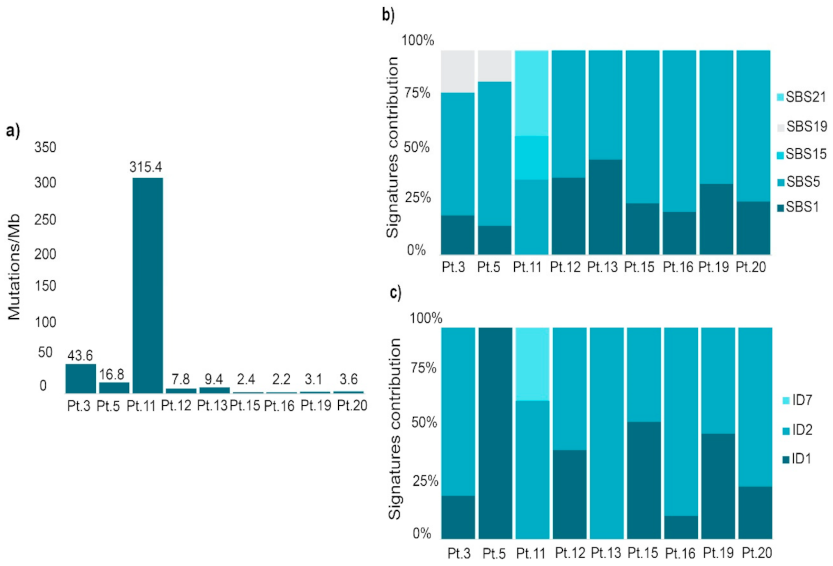
2.2. Mutational Profile Analysis
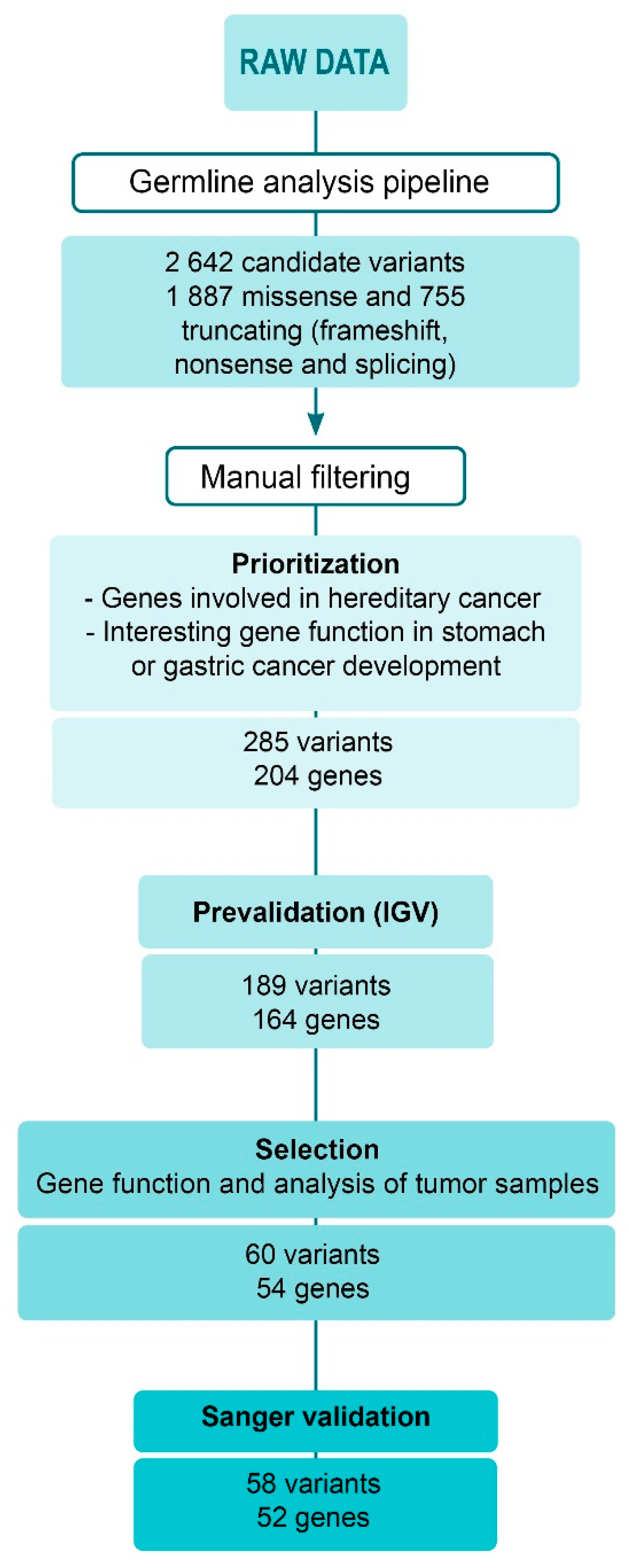
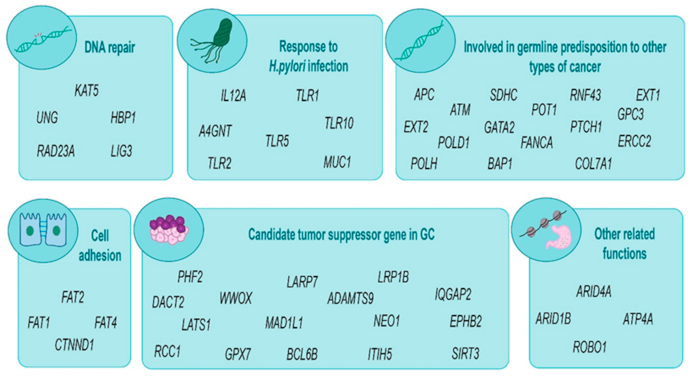
2.3. Germline Analysis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole-Exome Sequencing and Bioinformatic Analysis
4.3. Mutational Profile Analysis of Tumoral Samples
4.4. Variant Prioritization
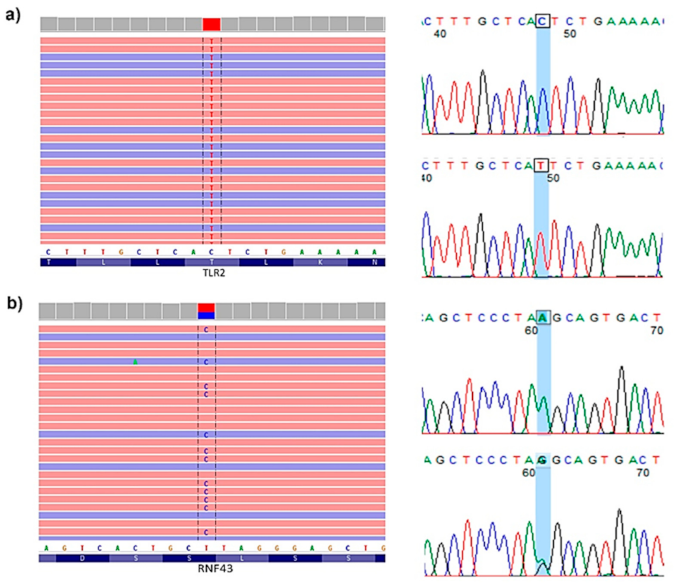
4.5. Variant Prevalidation, Selection and Final Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kluijt, I.; Sijmons, R.H.; Hoogerbrugge, N.; Plukker, J.T.; De Jong, D.; Van Krieken, J.H.; Van Hillegersberg, R.; Ligtenberg, M.; Bleiker, E.; Cats, A. Familial gastric cancer: Guidelines for diagnosis, treatment and periodic surveillance. Fam. Cancer 2012, 11, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt At a Histo-Clinical Classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Cubiella, J.; Pérez Aisa, Á.; Cuatrecasas, M.; Díez Redondo, P.; Fernández Esparrach, G.; Marín-Gabriel, J.C.; Moreira, L.; Núñez, H.; Pardo López, M.L.; Rodríguez de Santiago, E.; et al. Gastric cancer screening in low incidence populations: Position statement of AEG, SEED and SEAP. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Yusefi, A.R.; Lankarani, K.B.; Bastani, P.; Radinmanesh, M.; Kavosi, Z. Risk factors for gastric cancer: A systematic review. Asian Pac. J. Cancer Prev. 2018, 19, 591–603. [Google Scholar]
- Lott, P.C.; Carvajal-Carmona, L.G. Resolving gastric cancer aetiology: An update in genetic predisposition. Lancet Gastroenterol. Hepatol. 2018, 3, 874–883. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef]
- Majewski, I.J.; Kluijt, I.; Cats, A.; Scerri, T.S.; De Jong, D.; Kluin, R.J.C.; Hansford, S.; Hogervorst, F.B.L.; Bosma, A.J.; Hofland, I.; et al. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J. Pathol. 2013, 229, 621–629. [Google Scholar] [CrossRef]
- Li, J.; Woods, S.L.; Healey, S.; Beesley, J.; Chen, X.; Lee, J.S.; Sivakumaran, H.; Wayte, N.; Nones, K.; Waterfall, J.J.; et al. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am. J. Hum. Genet. 2016, 98, 830–842. [Google Scholar] [CrossRef]
- Milne, A.N.; Offerhaus, G.J.A. Early-onset gastric cancer: Learning lessons from the young. World J. Gastrointest. Oncol. 2010, 2, 59–64. [Google Scholar] [CrossRef]
- Machlowska, J.; Kapusta, P.; Baj, J.; Morsink, F.H.M.; Wołkow, P.; Maciejewski, R.; Johan, G.A.O.; Sitarz, R. High-Throughput Sequencing of Gastric Cancer Patients: Unravelling Genetic Predispositions Towards an Early-Onset Subtype. Cancers 2020, 12, 1981. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Roviello, F. Germline mutations of the E-cadherin gene (CDH1) in early onset gastric cancer. Semin. Oncol. 2020, 47, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; Mcpherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-C.; Nehoray, B.; Tao, S.; Castillo, D.; Woo, Y.; Sand, S.; Horcasitas, D.; Adamson, A.W.; Weitzel, J.; Wu, X.; et al. Genetic Gastric Cancer Susceptibility in the International Clinical Cancer Genomics Community Research Network. Cancer Genet. 2017, 216–217, 111–119. [Google Scholar]
- Sahasrabudhe, R.; Lott, P.; Bohorquez, M.; Toal, T.; Estrada, A.P.; Suarez, J.J.; Brea-Fernández, A.; Cameselle-Teijeiro, J.; Pinto, C.; Ramos, I.; et al. Germline Mutations in PALB2, BRCA1, and RAD51C, Which Regulate DNA Recombination Repair, in Patients With Gastric Cancer. Gastroenterology 2017, 152, 983–986.e6. [Google Scholar] [CrossRef]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: A whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef]
- Vogelaar, I.P.; Van Der Post, R.S.; Van Krieken, J.H.J.M.; Spruijt, L.; Van Zelst-Stams, W.A.G.; Kets, C.M.; Lubinski, J.; Jakubowska, A.; Teodorczyk, U.; Aalfs, C.M.; et al. Unraveling genetic predisposition to familial or early onset gastric cancer using germline whole-exome sequencing. Eur. J. Hum. Genet. 2017, 25, 1246–1252. [Google Scholar] [CrossRef]
- Weren, R.D.A.; Van Der Post, R.S.; Vogelaar, I.P.; Han Van Krieken, J.; Spruijt, L.; Lubinski, J.; Jakubowska, A.; Teodorczyk, U.; Aalfs, C.M.; Van Hest, L.P.; et al. Role of germline aberrations affecting CTNNA1, MAP3K6 and MYD88 in gastric cancer susceptibility. J. Med. Genet. 2018, 55, 669–674. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef]
- Cao, Z.; Xue, J.; Cheng, Y.; Wang, J.; Liu, Y.; Li, H.; Jiang, W.; Li, G. MDM2 promotes genome instability by ubiquitinating the transcription factor HBP1. Oncogene 2019, 4, 4835–4855. [Google Scholar] [CrossRef]
- Wang, H.; Shen, L.; Li, Y.; Lv, J. Integrated characterisation of cancer genes identifies key molecular biomarkers in stomach adenocarcinoma. J. Clin. Pathol. 2020, 73, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Feng, D.; Hu, L.; Chen, H.; Yang, G.; Cai, Q.; Gao, C.; Wei, D. FAT4 functions as a tumour suppressor in gastric cancer by modulating Wnt/β-catenin signalling. Br. J. Cancer 2015, 113, 1720–1729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rahman, N. Realising the Promise of Cancer Predisposition Genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Liang, X.; Chen, J.; Hong, J.; Li, L.; He, Q.; Cai, X. History and progression of fat cadherins in health and disease. OncoTargets Ther. 2016, 9, 7337–7343. [Google Scholar] [CrossRef]
- Katoh, M. Function and cancer genomics of FAT family genes (Review). Int. J. Oncol. 2012, 41, 1913–1918. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, Z.; Xia, Y.; Luo, J.; Xu, J.; He, X.; Tao, H. Low FAT4 expression is associated with a poor prognosis in gastric cancer patients. Oncotarget 2018, 9, 5137–5154. [Google Scholar] [CrossRef]
- Alders, M.; Al-Gazali, L.; Cordeiro, I.; Dallapiccola, B.; Garavelli, L.; Tuysuz, B.; Salehi, F.; Haagmans, M.A.; Mook, O.R.; Majoie, C.B.; et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum. Genet. 2014, 133, 1161–1167. [Google Scholar] [CrossRef]
- Thoreson, M.A.; Reynolds, A.B. Altered expression of the catenin p120 in human cancer: Implications for tumor progression. Differentiation 2002, 70, 583–589. [Google Scholar] [CrossRef]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of Epithelial Plasticity Determines Metastatic Organotropism in Pancreatic Cancer. Dev. Cell 2018, 45, 696–711.e8. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Leach, S.; Kaurah, P.; Jeyes, J.; Butterfield, Y.; Huntsman, D.; Brooks-Wilson, A.R. Catenin family genes are not commonly mutated in hereditary diffuse gastric cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2272–2274. [Google Scholar] [CrossRef]
- Ireton, R.C.; Davis, M.A.; Van Hengel, J.; Mariner, D.J.; Barnes, K.; Thoreson, M.A.; Anastasiadis, P.Z.; Matrisian, L.; Bundy, L.M.; Sealy, L.; et al. A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 2002, 159, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Söderberg, O.; Simões-Correia, J.; Grannas, K.; Suriano, G.; Seruca, R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur. J. Hum. Genet. 2013, 21, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Quin, S.-H.; Ruan, X.-Z.; Wang, X. p120-catenin participates in the progress of gastric cancer through regulating the Rac1 and Pak1 signaling pathway. Oncol. Rep. 2015, 34, 2357–2364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xing, A.Y.; Wang, Y.W.; Su, Z.X.; Shi, D.B.; Wang, B.; Gao, P. Catenin-δ 1, negatively regulated by miR-145, promotes tumour aggressiveness in gastric cancer. J. Pathol. 2015, 236, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cao, M.; Song, L.; Qi, P.; Chen, C.; Wang, X.; Li, N.; Peng, J.; Wu, D.; Hu, G.; et al. The contribution of toll-like receptor 2 on Helicobacter pylori activation of the nuclear factor-kappa B signaling pathway in gastric epithelial cells. Microb. Pathog. 2016, 98, 63–68. [Google Scholar] [CrossRef]
- Smith, S.M.; Moran, A.P.; Duggan, S.P.; Ahmed, S.E.; Mohamed, A.S.; Windle, H.J.; O’Neill, L.A.; Kelleher, D.P. Tribbles 3: A novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J. Immunol. 2011, 186, 2462–2471. [Google Scholar] [CrossRef]
- Yokota, S.-i.; Okabayashi, T.; Rehli, M.; Fujii, N.; Amano, K.-i. Helicobacter pylori lipopolysaccharides upregulate Toll-Like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect. Immun. 2010, 78, 468–476. [Google Scholar] [CrossRef]
- West, A.C.; Tang, K.; Tye, H.; Yu, L.; Deng, N.; Najdovska, M.; Lin, S.J.; Balic, J.J.; Okochi-Takada, E.; McGuirk, P.; et al. Identification of a TLR2-regulated gene signature associated with tumor cell growth in gastric cancer. Oncogene 2017, 36, 5134–5144. [Google Scholar] [CrossRef]
- Nemati, M.; Larussa, T.; Khorramdelazad, H.; Mahmoodi, M.; Jafarzadeh, A. Toll-like receptor 2: An important immunomodulatory molecule during Helicobacter pylori infection. Life Sci. 2017, 178, 17–29. [Google Scholar] [CrossRef]
- de Oliveira, J.G.; Silva, A.E. Polymorphisms of the TLR2 and TLR4 genes are associated with risk of gastric cancer in a Brazilian population. World J. Gastroenterol. 2012, 18, 1235–1242. [Google Scholar] [CrossRef]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.L.; Fock, K.M.; Mitchell, H.M. The Role of TLR2, TLR4 and CD14 genetic polymorphisms in gastric carcinogenesis: A case-control study and meta-analysis. PLoS ONE 2013, 8, e60327. [Google Scholar] [CrossRef] [PubMed]
- Saeki, N.; Saito, A.; Choi, I.J.; Matsuo, K.; Ohnami, S.; Totsuka, H.; Chiku, S.; Kuchiba, A.; Lee, Y.; Yoon, K.; et al. A functional single nucleotide polymorphism in Mucin 1, at chromosome 1q22, determines susceptibility to diffuse-type gastric cancer. Gastroenterology 2011, 140, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Liu, B.; Shrock, M.S.; Williams, T.; Aldaz, C.M. WWOX, the FRA16D gene: A target of and a contributor to genomic instability. Genes Chromosom. Cancer 2019, 58, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Xu, L.; Li, X.; Chu, E.S.H.; Zhao, G.; Go, M.Y.Y.; Tao, Q.; Jin, H.; Zeng, Z.; Sung, J.J.Y.; Yu, J. Epigenetic inactivation of BCL6B, a novel functional tumour suppressor for gastric cancer, is associated with poor survival. Gut 2012, 61, 977–985. [Google Scholar] [CrossRef]
- Cai, W.Y.; Lin, L.Y.; Wang, L.; Yang, L.; Ye, G.D.; Zeng, Q.; Cheng, J.; Xie, Y.Y.; Chen, M.L.; Luo, Q.C. Inhibition of Bcl6b promotes gastric cancer by amplifying inflammation in mice. Cell Commun. Signal. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, G.; Chu, S.-J.; Zhu, J.-S.; Zhang, R.; Lu, W.-W.; Xia, L.-Q.; Lu, Y.-M.; Da, W.; Sun, Q. Loss of large tumor suppressor 1 promotes growth and metastasis of gastric cancer cells through upregulation of the YAP signaling. Oncotarget 2016, 7, 16180–16193. [Google Scholar] [CrossRef]
- Chiurillo, M.A. Role of the Wnt/β-catenin pathway in gastric cancer: An in-depth literature review. World J. Exp. Med. 2015, 5, 84–102. [Google Scholar] [CrossRef]
- Pouptsis, A.; Swafe, L.; Patwardhan, M.; Stavraka, C. Surgical and Systemic Treatment of Hereditary Breast Cancer: A Mini-Review With a Focus on BRCA1 and BRCA2 Mutations. Front. Oncol. 2020, 10, 553080. [Google Scholar] [CrossRef]
- Llach, J.; Moreno, L.; Sánchez, A.; Herrera-Pariente, C.; Ocaña, T.; Cuatrecasas, M.; Rivero-Sánchez, L.; Moreira, R.; Díaz, M.; Jung, G.; et al. Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy. Cancers 2020, 12, 2386. [Google Scholar] [CrossRef]
- Llach, J.; Carballal, S.; Moreira, L. Familial pancreatic cancer: Current perspectives. Cancer Manag. Res. 2020, 12, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lu, Y. A review of capecitabine-based adjuvant therapy for gastric cancer in the Chinese population. Futur. Oncol. 2018, 14, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.F.; van Dijk, F.; de Boer, E.N.; van Dijk-Bos, K.K.; Jongbloed, J.D.H.; van der Hout, A.H.; Westers, H.; Sinke, R.J.; Swertz, M.A.; Sijmons, R.H.; et al. CoNVaDING: Single Exon Variation Detection in Targeted NGS Data. Hum. Mutat. 2016, 37, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at cancer diagnosis; median (IQR) | 41.5 (34.2–46.5) |
| Gender (women); number (%) | 13 (65) |
| Personal history of other neoplasm, number (%) | 0 |
| Family history of GC | |
| (FDR, SDR, TDR), number (%) | 5 (25) |
| Age at cancer diagnosis; median (range) | 61 (46–70) |
| Familial gastric cancer criteria, number (%) | 0 |
| Family history of other tumors (FDR, SDR, TDR), number (%) | 10 (50) |
| Pancreas | 3 (15) |
| Lung | 2 (10) |
| Brain | 1 (5) |
| Bone | 1 (5) |
| Leukemia | 1 (5) |
| Liver | 1 (5) |
| Endometrium | 1 (5) |
| Tumor location, number (%) | |
| Cardias | 1 (5) |
| Fundus | 3 (15) |
| Body | 13 (65) |
| Antrum | 3 (15) |
| Tumor stage, number (%) | |
| I/II | 13 (65) |
| III/IV | 7 (35) |
| GC histology, number (%) | |
| Diffuse | 16 (80) |
| Intestinal | 4 (20) |
| Tumor differentiation grade, number (%) * | |
| Intestinal histology (n = 4): | |
| High-grade (poorly differentiated) | 1 (25) |
| Low grade (well-moderately differentiated) | 3 (75) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera-Pariente, C.; Capó-García, R.; Díaz-Gay, M.; Carballal, S.; Muñoz, J.; Llach, J.; Sánchez, A.; Bonjoch, L.; Arnau-Collell, C.; Soares de Lima, Y.; et al. Identification of New Genes Involved in Germline Predisposition to Early-Onset Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 1310. https://doi.org/10.3390/ijms22031310
Herrera-Pariente C, Capó-García R, Díaz-Gay M, Carballal S, Muñoz J, Llach J, Sánchez A, Bonjoch L, Arnau-Collell C, Soares de Lima Y, et al. Identification of New Genes Involved in Germline Predisposition to Early-Onset Gastric Cancer. International Journal of Molecular Sciences. 2021; 22(3):1310. https://doi.org/10.3390/ijms22031310
Chicago/Turabian StyleHerrera-Pariente, Cristina, Roser Capó-García, Marcos Díaz-Gay, Sabela Carballal, Jenifer Muñoz, Joan Llach, Ariadna Sánchez, Laia Bonjoch, Coral Arnau-Collell, Yasmin Soares de Lima, and et al. 2021. "Identification of New Genes Involved in Germline Predisposition to Early-Onset Gastric Cancer" International Journal of Molecular Sciences 22, no. 3: 1310. https://doi.org/10.3390/ijms22031310
APA StyleHerrera-Pariente, C., Capó-García, R., Díaz-Gay, M., Carballal, S., Muñoz, J., Llach, J., Sánchez, A., Bonjoch, L., Arnau-Collell, C., Soares de Lima, Y., Golubicki, M., Jung, G., Lozano, J. J., Castells, A., Balaguer, F., Bujanda, L., Castellví-Bel, S., & Moreira, L. (2021). Identification of New Genes Involved in Germline Predisposition to Early-Onset Gastric Cancer. International Journal of Molecular Sciences, 22(3), 1310. https://doi.org/10.3390/ijms22031310