
Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels



Abstract
1. Introduction
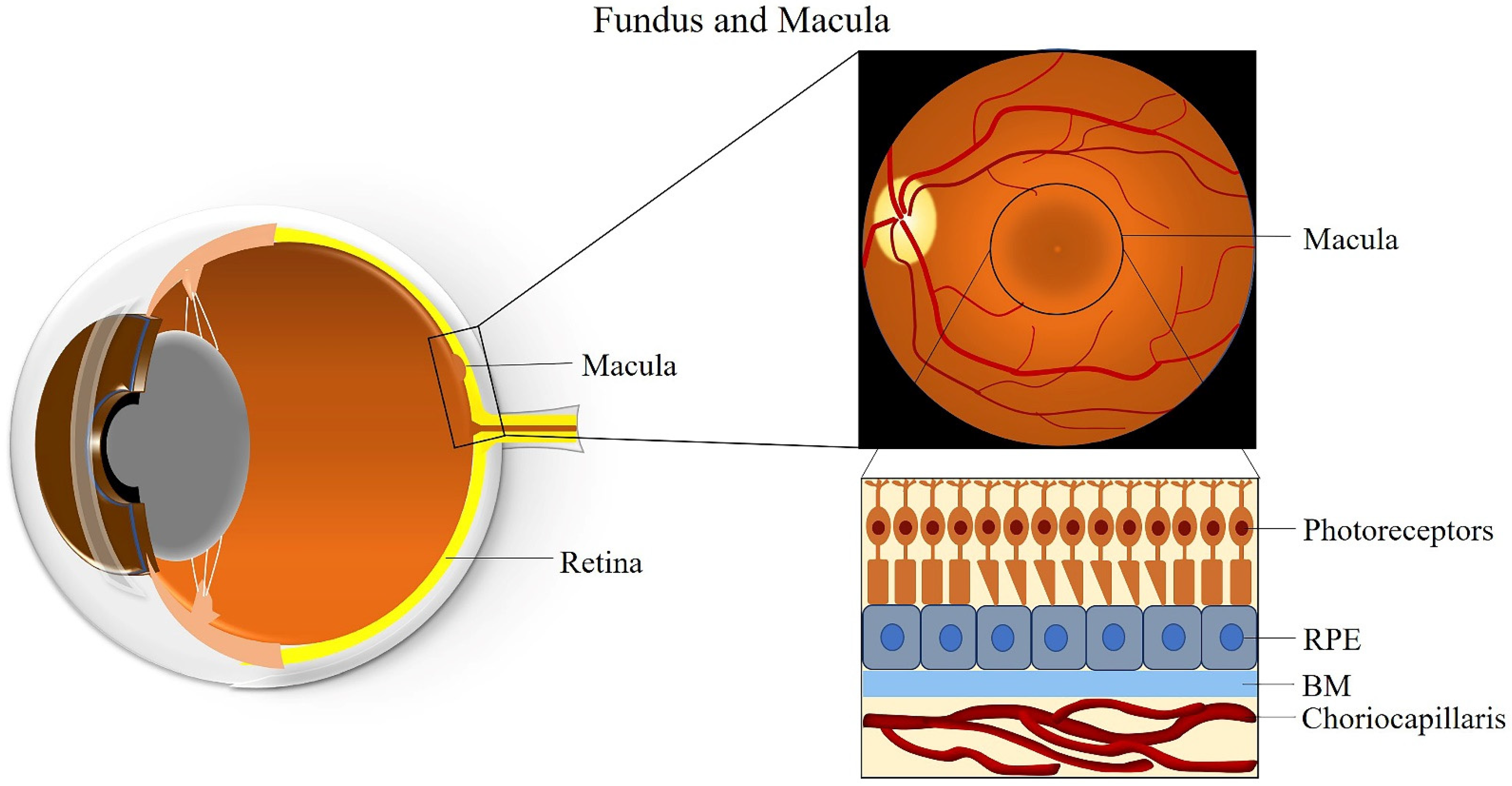
2. The Macula
3. Clinical Classification of AMD
4. Pathogenesis of AMD
4.1. Oxidative Stress and AMD
4.1.1. The Macula—An Ideal Environment for the Generation of ROS
4.1.2. Animal Models of AMD
4.1.3. The Generation of ROS Due to Light Exposure
4.1.4. Generation of ROS by Dysregulated Autophagy
4.1.5. Oxidative Stress and Disease Development
4.1.6. Genetics Involved in AMD
4.2. Choroidal Vascular Dysfunction and AMD
4.2.1. Choroidal Vascular Changes in AMD
4.2.2. The Mechanism Underlying CNV in AMD
4.3. ROS and Choroidal Vascular Dysfunction
5. Therapy Strategies in AMD Targeting the Vasculature and Oxidative Stress
5.1. Current Therapies
5.2. Novel Gene Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (amd): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Adelson, J.D.; Bourne, R.R.A. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to vision 2020: The right to sight: An analysis for the global burden of disease study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Ke, K.M.; Chakravarthy, U. Economic cost of age-related macular degeneration. Drugs Aging 2006, 23, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Cheung, C.M.G. Updates on the epidemiology of age-related macular degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar]
- Wong, W.L.; Su, X. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Brown, M.M.; Brown, G.C. Age-related macular degeneration: Economic burden and value-based medicine analysis. Can. J. Ophthalmol. 2005, 40, 277–287. [Google Scholar] [CrossRef]
- Donders, F.C. Beiträge zur pathologischen anatomie des auges. Arch. Für Ophthalmol. 1855, 1, 106–118. [Google Scholar] [CrossRef]
- Nagel, A. Ueber chorioiditis areolaris und über krystalle im augenhintergrund. Areolar Choroiditis Cryst. Eye Fundus. Klin Monbl Augenheilk 1868, 4, 417–420. [Google Scholar]
- De Jong, P.T.V.M. A historical analysis of the quest for the origins of aging macula disorder, the tissues involved, and its terminology: Supplementary issue: Ophthalmic history. Ophthalmol. Eye Dis. 2016, 8. [Google Scholar] [CrossRef]
- Haider, N.B.; Cruz, N.M. Pathobiology of the outer retina: Genetic and nongenetic causes of disease. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 2084–2114. [Google Scholar]
- Prevention and Treatment of Age-related Macular Degeneration. Available online: https://www.prescriber.co.uk/article/prevention-treatment-age-related-macular-degeneration/ (accessed on 14 November 2020).
- Lim, L.S.; Mitchell, P. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Bowes Rickman, C.; Farsiu, S. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Wong, T.Y. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Augood, C. Cigarette smoking and age-related macular degeneration in the eureye study. Ophthalmology 2007, 114, 1157–1163. [Google Scholar] [CrossRef]
- Cruickshanks, K.J.; Klein, R. Sunlight and the 5-year incidence of early age-related maculopathy: The beaver dam eye study. Arch. Ophthalmol. 2001, 119, 246–250. [Google Scholar]
- Tien, P.-T.; Lin, H.-J. Perfluorooctanoic acid in indoor particulate matter triggers oxidative stress and inflammation in corneal and retinal cells. Sci. Rep. 2020, 10, 15702. [Google Scholar] [CrossRef]
- Bellezza, I. Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef]
- Lipecz, A.; Miller, L. Microvascular contributions to age-related macular degeneration (amd): From mechanisms of choriocapillaris aging to novel interventions. GeroScience 2019, 41, 813–845. [Google Scholar] [CrossRef]
- Rakoczy, P.E.; Zhang, D. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am. J. Pathol. 2002, 161, 1515–1524. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.-H. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Archer, D.; Gardiner, T.J.A.j.o.o. Electron microscopic features of experimental choroidal neovascularization. Am. J. Ophthalmol. 1981, 91, 433–457. [Google Scholar] [CrossRef]
- Stone, E.M.; Braun, T.A. Missense variations in the fibulin 5 gene and age-related macular degeneration. N. Engl. J. Med. 2004, 351, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.Y.; Hartvigsen, K. Oxidation-specific epitopes are important targets of innate immunity. J. Intern. Med. 2008, 263, 479–488. [Google Scholar] [CrossRef] [PubMed]
- The Anatomy of the Macula. Available online: https://www.verywellhealth.com/macula-anatomy-function-and-significance-4771995 (accessed on 15 November 2020).
- Margolis, R.; Spaide, R.F. A pilot study of enhanced depth imaging optical coherence tomography of the choroid in normal eyes. Am. J. Ophthalmol. 2009, 147, 811–815. [Google Scholar] [CrossRef] [PubMed]
- The Macula. Available online: https://www.imaios.com/en/e-Anatomy/Anatomical-Parts/Macula (accessed on 27 January 2021).
- Tschulakow, A.V.; Oltrup, T. The anatomy of the foveola reinvestigated. PeerJ 2018, 6, e4482. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Sloan, K.R. Human photoreceptor topography. J. Comp. Neurol. 1990, 292, 497–523. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Hicks, D. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Hughes, B.A.; Gallemore, R.P.; Miller, S.S. The Retinal Pigment Epithelium: Current Aspects of Function and Disease; Marmor, M.F., Wolfensberger, T.J., Eds.; Oxford University Press: New York, NY, USA, 1998; pp. 103–134. [Google Scholar]
- Handa, J.T. How does the macula protect itself from oxidative stress? Mol. Asp. Med. 2012, 33, 418–435. [Google Scholar] [CrossRef]
- Fryczkowski, A.W. Anatomical and functional choroidal lobuli. Int. Ophthalmol. 1994, 18, 131–141. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Bailey, T.A. Bruch’s membrane and the vascular intima: Is there a common basis for age-related changes and disease? Clin. Exp. Ophthalmol. 2005, 33, 518–523. [Google Scholar] [CrossRef]
- Okubo, A.; Rosa, R.H., Jr. The relationships of age changes in retinal pigment epithelium and bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 443–449. [Google Scholar]
- Davis, M.D.; Gangnon, R.E. The age-related eye disease study severity scale for age-related macular degeneration: Areds report no. 17. Arch. Ophthalmol. 2005, 123, 1484–1498. [Google Scholar]
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-related eye disease study report number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar]
- Mitchell, P.; Foran, S. Age-related eye disease study severity scale and simplified severity scale for age-related macular degeneration. Arch. Ophthalmol. 2005, 123, 1598–1599. [Google Scholar] [CrossRef] [PubMed]
- Ferris, F.L.; Davis, M.D. A simplified severity scale for age-related macular degeneration: Areds report no. 18. Arch. Ophthalmol. 2005, 123, 1570–1574. [Google Scholar] [PubMed]
- Ferris, F.L.; Wilkinson, C.P. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W. Reactive oxygen species (ros) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Sun, Y.; Smith, L.E.H. Retinal vasculature in development and diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef]
- Winkler, B.S.; Boulton, M.E. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32. [Google Scholar]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.V.; Liles, M.R. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell. Res. 1994, 214, 242–249. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Gottlieb, E. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem. Photobiol. 2004, 79, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, M.E.; Neuringer, M. Animal models of age-related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [PubMed]
- Chader, G.J. Animal models in research on retinal degenerations: Past progress and future hope. Vis. Res. 2002, 42, 393–399. [Google Scholar] [CrossRef]
- Duncan, J.L.; LaVail, M.M. Intense cyclic light-induced retinal degeneration in rats. Arch. Ophthalmol. 2010, 128, 244–245. [Google Scholar] [CrossRef][Green Version]
- Wielgus, A.; Collier, R. Blue light induced a2e oxidation in rat eyes–experimental animal model of dry amd. Photochem. Photobiol. Sci. 2010, 9, 1505–1512. [Google Scholar] [CrossRef]
- Wenzel, A.; Grimm, C. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.-H. Central role of oxidative stress in age-related macular degeneration: Evidence from a review of the molecular mechanisms and animal models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J. Loss of nrf-2 and pgc-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Justilien, V.; Pang, J.-J. Sod2 knockdown mouse model of early amd. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Ding, J.-D. Pparβ/δ selectively regulates phenotypic features of age-related macular degeneration. Aging 2016, 8, 1952–1978. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Ohta, S. Blue light-induced oxidative stress in live skin. Free Radic. Biol. Med. 2017, 108, 300–310. [Google Scholar] [CrossRef]
- Marek, V.; Mélik-Parsadaniantz, S. Blue light phototoxicity toward human corneal and conjunctival epithelial cells in basal and hyperosmolar conditions. Free Radic. Biol. Med. 2018, 126, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Chalam, K.V.; Khetpal, V. A review: Role of ultraviolet radiation in age-related macular degeneration. Eye Contact Lens 2011, 37, 225–232. [Google Scholar] [CrossRef]
- Funk, R.H.; Schumann, U. Blue light induced retinal oxidative stress: Implications for macular degeneration. World J. Ophthalmol. 2014, 4, 29–34. [Google Scholar] [CrossRef]
- Glickman, R.D. Ultraviolet phototoxicity to the retina. Eye Contact Lens 2011, 37, 196–205. [Google Scholar] [CrossRef]
- Roehlecke, C.; Schumann, U. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef]
- Grimm, C.; Wenzel, A. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Investig. Ophthalmol. Vis. Sci. 2001, 42, 497–505. [Google Scholar]
- Sparrow, J.R.; Nakanishi, K. The lipofuscin fluorophore a2e mediates blue light–induced damage to retinal pigmented epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1981–1989. [Google Scholar]
- Dontsov, A.E.; Glickman, R.D. Retinal pigment epithelium pigment granules stimulate the photo-oxidation of unsaturated fatty acids. Free Radic. Biol. Med. 1999, 26, 1436–1446. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Boulton, M. Rpe lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A.J.F.R.B. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Shamsi, F.A.; Boulton, M. Inhibition of rpe lysosomal and antioxidant activity by the age pigment lipofuscin. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3041–3046. [Google Scholar]
- Ng, K.-P.; Gugiu, B. Retinal pigment epithelium lipofuscin proteomics. Mol. Cell. Proteom. 2008, 7, 1397–1405. [Google Scholar] [CrossRef]
- Kim, S.; Piri, N. Autophagy in the retina. Investig. Ophthalmol. Vis. Sci. 2007, 48, 55. [Google Scholar]
- Klionsky, D.J.; Abdelmohsen, K. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Naso, F.; Intartaglia, D. Light-responsive microrna mir-211 targets ezrin to modulate lysosomal biogenesis and retinal cell clearance. EMBO J. 2020, 39, e102468. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Bao, X.-L. Autophagy in age-related macular degeneration: A regulatory mechanism of oxidative stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C. Dysregulated autophagy in the rpe is associated with increased susceptibility to oxidative stress and amd. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science. 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Wang, L. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in rpe cells. Free Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cano, M. P62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim. Biophys. Acta 2014, 1843, 1248–1258. [Google Scholar] [CrossRef]
- Hanus, J.; Zhang, H. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Anderson, C. Rpe necroptosis in response to oxidative stress and in amd. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Lin, H. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Van Houten, B. Hydrogen peroxide causes significant mitochondrial DNA damage in human rpe cells. Exp. Eye Res. 1999, 68, 765–772. [Google Scholar] [CrossRef]
- Westphal, D.; Kluck, R.M. Building blocks of the apoptotic pore: How bax and bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014, 21, 196–205. [Google Scholar] [CrossRef]
- Li, G.-Y.; Fan, B. Calcium overload is a critical step in programmed necrosis of arpe-19 cells induced by high-concentration h2o2. Biomed. Environ. Sci. 2010, 23, 371–377. [Google Scholar] [CrossRef]
- Jin, G.-F.; Hurst, J.S. Hydrogen peroxide stimulates apoptosis in cultured human retinal pigment epithelial cells. Curr. Eye Res. 2001, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; DeWeerd, A.J. Mitochondrial oxidative stress in the retinal pigment epithelium (rpe) led to metabolic dysfunction in both the rpe and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Ferrington, D.A. Perspective on amd pathobiology: A bioenergetic crisis in the rpe. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kass, G.E.N. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E. Oxidative stress is the principal contributor to inflammasome activation in retinal pigment epithelium cells with defunct proteasomes and autophagy. Cell. Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef]
- Wang, K.; Yao, Y. Amyloid β induces nlrp3 inflammasome activation in retinal pigment epithelial cells via nadph oxidase- and mitochondria-dependent ros production. J. Biochem. Mol. Toxicol. 2017, 31, e21887. [Google Scholar] [CrossRef]
- Hong, P.; Gu, R.-N. Nlrp3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J. Neuroinflammation 2019, 16, 121. [Google Scholar] [CrossRef]
- Tseng, W.A.; Thein, T. Nlrp3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Chen, M.; Chan, C.C. Cholesterol homeostasis, macrophage malfunction and age-related macular degeneration. Ann Transl Med. 2018, 6, S55. [Google Scholar] [CrossRef]
- Duncan, K.G.; Hosseini, K. Expression of reverse cholesterol transport proteins atp-binding cassette a1 (abca1) and scavenger receptor bi (sr-bi) in the retina and retinal pigment epithelium. Br. J. Ophthalmol. 2009, 93, 1116–1120. [Google Scholar] [CrossRef]
- Ishida, B.Y.; Duncan, K.G. High density lipoprotein mediated lipid efflux from retinal pigment epithelial cells in culture. Br. J. Ophthalmol. 2006, 90, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Storti, F.; Raphael, G. Regulated efflux of photoreceptor outer segment-derived cholesterol by human rpe cells. Exp. Eye Res. 2017, 165, 65–77. [Google Scholar] [CrossRef]
- Biswas, L.; Zhou, X. Retinal pigment epithelium cholesterol efflux mediated by the 18 kda translocator protein, tspo, a potential target for treating age-related macular degeneration. Hum. Mol. Genet. 2017, 26, 4327–4339. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, K.B.; Handa, J.T. Lipids, lipoproteins, and age-related macular degeneration. J. Lipids 2011, 2011, 802059. [Google Scholar] [CrossRef]
- Li, C.M.; Clark, M.E. Apolipoprotein localization in isolated drusen and retinal apolipoprotein gene expression. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaft, T.L.; de Bruijn, W.C. Is basal laminar deposit unique for age-related macular degeneration? Arch. Ophthalmol. 1991, 109, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Finnemann, S.C.; Leung, L.W. The lipofuscin component a2e selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 3842–3847. [Google Scholar] [CrossRef] [PubMed]
- Paun, C.C.; Ersoy, L. Genetic variants and systemic complement activation levels are associated with serum lipoprotein levels in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7766–7773. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Heidmann, D.G.; Sall, J. Basal laminar deposit formation in apo b100 transgenic mice: Complex interactions between dietary fat, blue light, and vitamin E. Investig. Ophthalmol. Vis. Sci. 2004, 45, 260–266. [Google Scholar] [CrossRef]
- Mares-Perlman, J.A.; Brady, W.E. Dietary fat and age-related maculopathy. Arch. Ophthalmol. 1995, 113, 743–748. [Google Scholar] [CrossRef]
- Jun, S.; Datta, S. The impact of lipids, lipid oxidation, and inflammation on amd, and the potential role of mirnas on lipid metabolism in the rpe. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. The important role of lipid peroxidation processes in aging and age dependent diseases. Mol. Biotechnol. 2007, 37, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. Peroxyl radicals: Inductors of neurodegenerative and other inflammatory diseases. Their origin and how they transform cholesterol, phospholipids, plasmalogens, polyunsaturated fatty acids, sugars, and proteins into deleterious products. Free Radic. Biol. Med. 2006, 41, 362–387. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, T.; Spangler, R. The role of lox and loxl2 in the pathogenesis of an experimental model of choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5280–5289. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Ahmad, S. 12/15-lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: Contribution of nadph oxidase. PLoS ONE 2013, 8, e57254. [Google Scholar] [CrossRef]
- Subramanian, P.; Mendez, E.F. A novel inhibitor of 5-lipoxygenase (5-lox) prevents oxidative stress-induced cell death of retinal pigment epithelium (rpe) cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4581–4588. [Google Scholar] [CrossRef]
- Yadav, U.C.S.; Ramana, K.V. Regulation of nf-κb-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell. Longev. 2013, 2013, 690545. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kapphahn, R.J. Increased retinal mtdna damage in the cfh variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef]
- Jones, M.M.; Manwaring, N. Mitochondrial DNA haplogroups and age-related maculopathy. Arch. Ophthalmol. 2007, 125, 1235–1240. [Google Scholar] [CrossRef]
- Canter, J.A.; Olson, L.M. Mitochondrial DNA polymorphism a4917g is independently associated with age-related macular degeneration. PLoS ONE 2008, 3, e2091. [Google Scholar] [CrossRef]
- Haines, J.L.; Hauser, M.A. Complement factor h variant increases the risk of age-related macular degeneration. Science 2005, 308, 419. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.M.; Mott, S.L. Complement-regulatory proteins cfhr1 and cfhr3 and patient response to anti-cd20 monoclonal antibody therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 954–961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fritsche, L.G.; Chen, W. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [PubMed]
- Fagerness, J.A.; Maller, J.B. Variation near complement factor i is associated with risk of advanced amd. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Lauer, N. An imbalance of human complement regulatory proteins cfhr1, cfhr3 and factor h influences risk for age-related macular degeneration (amd). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Klaver, C.C.W.; Kliffen, M. Genetic association of apolipoprotein e with age-related macular degeneration. Am. J. Hum. Genet. 1998, 63, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Cezario, S.M.; Calastri, M.C.J. Association of high-density lipoprotein and apolipoprotein e genetic variants with age-related macular degeneration. Arq. Bras. Oftalmol. 2015, 78, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Dithmar, S.; Curcio, C.A. Ultrastructural changes in bruch’s membrane of apolipoprotein e–deficient mice. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2035–2042. [Google Scholar]
- Zadeh, J.K.; Zhutdieva, M.B. Apolipoprotein e deficiency causes endothelial dysfunction in the mouse retina. Oxid. Med. Cell. Longev. 2019, 2019, 5181429. [Google Scholar] [CrossRef]
- Malek, G.; Johnson, L.V. Apolipoprotein e allele-dependent pathogenesis: A model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 11900–11915. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Stambolian, D. Genetic variants near TIMP3 and high-density lipoprotein–associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Fagerness, J. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, M.; Winkler, B. Increased expression of vascular endothelial growth factor associated with accumulation of lipids in bruch’s membrane of ldl receptor knockout mice. Br. J. Ophthalmol. 2005, 89, 1627. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zerbib, J.; Seddon, J.M. Rs5888 variant of scarb1 gene is a possible susceptibility factor for age-related macular degeneration. PLoS ONE 2009, 4, e7341. [Google Scholar] [CrossRef]
- Von Rückmann, A.; Fitzke, F.W. Fundus autofluorescence in age-related macular disease imaged with a laser scanning ophthalmoscope. Investig. Ophthalmol. Vis. Sci. 1997, 38, 478–486. [Google Scholar]
- Wu, Z.; Luu, C.D. Optical coherence tomography-defined changes preceding the development of drusen-associated atrophy in age-related macular degeneration. Ophthalmology 2014, 121, 2415–2422. [Google Scholar] [CrossRef]
- Biesemeier, A.; Taubitz, T. Choriocapillaris breakdown precedes retinal degeneration in age-related macular degeneration. Neurobiol. Aging 2014, 35, 2562–2573. [Google Scholar] [CrossRef]
- Von der Emde, L.; Thiele, S. Assessment of exudative activity of choroidal neovascularization in age-related macular degeneration by oct angiography. Ophthalmologica 2020, 243, 120–128. [Google Scholar] [CrossRef]
- De Carlo, T.E.; Romano, A. A review of optical coherence tomography angiography (octa). Int. J. Retin. Vitr. 2015, 1, 5. [Google Scholar] [CrossRef]
- Wong, T.; Chakravarthy, U. The natural history and prognosis of neovascular age-related macular degeneration: A systematic review of the literature and meta-analysis. Ophthalmology 2008, 115, 116–126.e1. [Google Scholar] [CrossRef] [PubMed]
- Sulzbacher, F.; Pollreisz, A. Identification and clinical role of choroidal neovascularization characteristics based on optical coherence tomography angiography. Acta Ophthalmol. 2017, 95, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Farazdaghi, M.K.; Ebrahimi, K.B. Role of the choroid in age-related macular degeneration: A current review. J. Ophthalmic Vis. Res. 2019, 14, 78–87. [Google Scholar] [PubMed]
- Nagiel, A.; Bansal, M. Morphological analysis of type 1, type 2, and type 3 neovascularization in exudative age-related macular degeneration using oct angiography. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3965. [Google Scholar]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (amd): Relationships between the photoreceptor/retinal pigment epithelium/bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef]
- Kwak, N.; Okamoto, N. Vegf is major stimulator in model of choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3158–3164. [Google Scholar]
- Yi, X.; Ogata, N. Vascular endothelial growth factor expression in choroidal neovascularization in rats. Graefe’s Arch. Clin. Exp. Ophthalmol. 1997, 235, 313–319. [Google Scholar] [CrossRef]
- Kvanta, A.; Algvere, P.V. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1929–1934. [Google Scholar] [CrossRef]
- Blaauwgeers, H.G.T.; Holtkamp, G.M. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris: Evidence for a trophic paracrine relation. Am. J. Pathol. 1999, 155, 421–428. [Google Scholar] [CrossRef]
- Spilsbury, K.; Garrett, K.L. Overexpression of vascular endothelial growth factor (vegf) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol. 2000, 157, 135–144. [Google Scholar] [CrossRef]
- Liu, S.; Biesemeier, A.K. A new rat model of treatment-naive quiescent choroidal neovascularization induced by human vegf165 overexpression. Biol. Open 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ramshekar, A. Iqgap1 causes choroidal neovascularization by sustaining vegfr2-mediated rac1 activation. Angiogenesis 2020, 23, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Testini, C.; Smith, R.O. Myc-dependent endothelial proliferation is controlled by phosphotyrosine 1212 in vegf receptor-2. EMBO Rep. 2019, 20, e47845. [Google Scholar] [CrossRef] [PubMed]
- Monaghan-Benson, E.; Hartmann, J. The role of vascular endothelial growth factor-induced activation of nadph oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef]
- Marneros, A.G.; Fan, J. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am. J. Pathol. 2005, 167, 1451–1459. [Google Scholar] [CrossRef]
- Yeo, N.J.Y.; Chan, E.J.J. Choroidal neovascularization: Mechanisms of endothelial dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T.J.C. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Heiss, C.; Rodriguez-Mateos, A. Central role of enos in the maintenance of endothelial homeostasis. Antioxid. Redox Signal. 2015, 22, 1230–1242. [Google Scholar] [CrossRef]
- Gericke, A.; Wolff, I. Retinal arteriole reactivity in mice lacking the endothelial nitric oxide synthase (enos) gene. Exp. Eye Res. 2019, 181, 150–156. [Google Scholar] [CrossRef]
- Laspas, P.; Goloborodko, E. Role of nitric oxide synthase isoforms for ophthalmic artery reactivity in mice. Exp. Eye Res. 2014, 127, 1–8. [Google Scholar] [CrossRef]
- Albrecht, E.W.J.A.; Stegeman, C.A. Protective role of endothelial nitric oxide synthase. J. Pathol. 2003, 199, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ferrige, A.G. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- McDougal, D.H.; Gamlin, P.D. Autonomic control of the eye. Compr. Physiol. 2015, 5, 439–473. [Google Scholar] [PubMed]
- Kashiwagi, S.; Kajimura, M. Nonendothelial source of nitric oxide in arterioles but not in venules: Alternative source revealed in vivo by diaminofluorescein microfluorography. Circ. Res. 2002, 91, e55–e64. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Baba, T. Low nitric oxide synthases (nos) in eyes with age-related macular degeneration (amd). Exp. Eye Res. 2009, 90, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Hattenbach, L.O.; Falk, B. Detection of inducible nitric oxide synthase and vascular endothelial growth factor in choroidal neovascular membranes. Ophthalmologica 2002, 216, 209–214. [Google Scholar] [CrossRef]
- Ando, A.; Yang, A. Blockade of nitric-oxide synthase reduces choroidal neovascularization. Mol. Pharmacol. 2002, 62, 539. [Google Scholar] [CrossRef]
- Jiang, H.; Wu, M. Serine racemase deficiency attenuates choroidal neovascularization and reduces nitric oxide and vegf levels by retinal pigment epithelial cells. J. Neurochem. 2017, 143, 375–388. [Google Scholar] [CrossRef]
- Pechánová, O.; Simko, F. The role of nitric oxide in the maintenance of vasoactive balance. Physiol. Res. Acad. Sci. Bohemoslov. 2007, 56, S7–S16. [Google Scholar]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of nadph oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Z. Endothelial nadph oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar] [PubMed]
- Chen, C.C.; Chow, M.P. Flavonoids inhibit tumor necrosis factor-alpha-induced up-regulation of intercellular adhesion molecule-1 (icam-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-kappab: Structure-activity relationships. Mol. Pharm. 2004, 66, 683–693. [Google Scholar]
- Nagata, M. Inflammatory cells and oxygen radicals. Curr. Drug Targets Inflamm. Allergy 2005, 4, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabrawey, M.; Rojas, M. Role of nadph oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Mei, A. Nadph oxidase 4 mediates insulin-stimulated hif-1α and vegf expression, and angiogenesis in vitro. PLoS ONE 2012, 7, e48393. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H. Important role of nox4 type nadph oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Kannan, R. Thiol regulation of vascular endothelial growth factor-a and its receptors in human retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2006, 346, 1200–1206. [Google Scholar] [CrossRef]
- Wang, H.; Geisen, P. The role of rpe cell-associated vegf189 in choroidal endothelial cell transmigration across the rpe. Investig. Ophthalmol. Vis. Sci. 2011, 52, 570–578. [Google Scholar] [CrossRef]
- Li, Q.; Dinculescu, A. Downregulation of p22phox in retinal pigment epithelial cells inhibits choroidal neovascularization in mice. Mol. Ther. 2008, 16, 1688–1694. [Google Scholar] [CrossRef]
- Ushio–Fukai, M. Vegf signaling through nadph oxidase-derived ros. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Fleissner, F.; Thum, T. Critical role of the nitric oxide/reactive oxygen species balance in endothelial progenitor dysfunction. Antioxid. Redox Signal. 2011, 15, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Gori, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Daiber, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cgmp-dependent protein kinase. Arter. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Redox signaling in cardiovascular health and disease. Free Radic. Biol. Med. 2013, 61, 473–501. [Google Scholar] [CrossRef]
- Xu, C.; Tang, F. Astragaloside iv improves the isoproterenol-induced vascular dysfunction via attenuating enos uncoupling-mediated oxidative stress and inhibiting ros-nf-κb pathways. Int. Immunopharmacol. 2016, 33, 119–127. [Google Scholar] [CrossRef]
- Marumo, T.; Noll, T. Significance of nitric oxide and peroxynitrite in permeability changes of the retinal microvascular endothelial cell monolayer induced by vascular endothelial growth factor. J. Vasc. Res. 1999, 36, 510–515. [Google Scholar] [CrossRef]
- Khandhadia, S.; Lotery, A. Oxidation and age-related macular degeneration: Insights from molecular biology. Expert Rev. Mol. Med. 2010, 12, e34. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 7. [Google Scholar] [CrossRef]
- Singh, S.; Aggarwal, B.B.J.J.o.B.C. Activation of transcription factor nf-κb is suppressed by curcumin (diferuloylmethane). J. Biol. Chem. 1995, 270, 24995–25000. [Google Scholar] [CrossRef]
- Wu, J.; Cho, E. Intakes of lutein, zeaxanthin, and other carotenoids and age-related macular degeneration during 2 decades of prospective follow-up. JAMA Ophthalmol. 2015, 133, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Beyond lipid lowering: The role of statins in vascular protection. Int. J. Cardiol. 2002, 86, 5–18. [Google Scholar] [CrossRef]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mrna stability by rho gtpase*. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef]
- Wagner, A.H.; Köhler, T. Improvement of nitric oxide-dependent vasodilatation by hmg-coa reductase inhibitors through attenuation of endothelial superoxide anion formation. Arter. Thromb. Vasc. Biol. 2000, 20, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Chen, X.-J.J. Simvastatin inhibits interleukin-6 release in human monocytes stimulated by c-reactive protein and lipopolysaccharide. Coron. Artery Dis. 2003, 14, 329–334. [Google Scholar] [CrossRef]
- Guymer, R.H.; Chiu, A.W. Hmg coa reductase inhibitors (statins): Do they have a role in age-related macular degeneration? Surv. Ophthalmol. 2005, 50, 194–206. [Google Scholar] [CrossRef]
- Curcio, C.A.; Messinger, J.D. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: Morphology, prevalence, topography, and biogenesis model. Retina 2013, 33, 265–276. [Google Scholar] [CrossRef]
- De Sotomayor, M.a.Á.; Andriantsitohaina, R.J.B. Simvastatin and ca2+ signaling in endothelial cells: Involvement of rho protein. Biochem. Biophys. Res. Commun. 2001, 280, 486–490. [Google Scholar] [CrossRef]
- Tsujinaka, H.; Itaya-Hironaka, A. Statins decrease vegf expression in retinal pigment epithelial cells by downregulation of receptor for age (rage). Am. Diabetes Assoc. 2018. [Google Scholar] [CrossRef]
- Advances in Therapy for Dry Amd. Available online: https://www.oraclinical.com/resource/advances-in-therapy-for-dry-amd/ (accessed on 29 December 2020).
- Al-Holou, S.N.; Tucker, W.R. The association of statin use with age-related macular degeneration progression: The age-related eye disease study 2 report number 9. Ophthalmology 2015, 122, 2490–2496. [Google Scholar] [CrossRef]
- Gehlbach, P.; Li, T. Statins for age-related macular degeneration. Cochrane Database Syst. Rev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- AnandBabu, K.; Bharathidevi, S.R. Serum paraoxonase activity in relation to lipid profile in age-related macular degeneration patients. Exp. Eye Res. 2016, 152, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.; Wang, J.J. Dietary fatty acids and the 10-year incidence of age-related macular degeneration: The blue mountains eye study. Arch. Ophthalmol. 2009, 127, 656–665. [Google Scholar] [CrossRef]
- Tan, J.S.L.; Mitchell, P. Statins and the long-term risk of incident age-related macular degeneration: The blue mountains eye study. Am. J. Ophthalmol. 2007, 143, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, Y. The association between statin use and risk of age-related macular degeneration. Sci. Rep. 2015, 5, 18280. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.J.F. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Korani, S.; Korani, M. Application of nanotechnology to improve the therapeutic benefits of statins. Drug Discov. Today 2018, 24, 567–574. [Google Scholar] [CrossRef]
- Yadav, M.; Schiavone, N. Atorvastatin-loaded solid lipid nanoparticles as eye drops: Proposed treatment option for age-related macular degeneration (amd). Drug Deliv. Transl. Res. 2020, 10, 919–944. [Google Scholar] [CrossRef]
- Markham, A. Brolucizumab: First approval. Drugs 2019, 79, 1997–2000. [Google Scholar] [CrossRef]
- Anti-Vegf Treatments for Wet Age-related Macular Degeneration. Available online: https://www.brightfocus.org/macular/article/anti-vegf-treatments-wet-age-related-macular (accessed on 1 December 2020).
- Stieger, K.; Cronin, T. Adeno-associated virus mediated gene therapy for retinal degenerative diseases. Methods Mol. Biol. 2011, 807, 179–218. [Google Scholar]
- Prea, S.M.; Chan, E.C. Gene therapy with endogenous inhibitors of angiogenesis for neovascular age-related macular degeneration: Beyond anti-vegf therapy. J. Ophthalmol. 2015, 2015, 201726. [Google Scholar] [CrossRef] [PubMed]
- Gene Therapy Discovery Offers Hope for Patients with Amd. Available online: https://www.genengnews.com/news/gene-therapy-discovery-offers-hope-for-patients-with-amd/ (accessed on 1 December 2020).
- Akyol, E.; Lotery, A. Gene, cell and antibody-based therapies for the treatment of age-related macular degeneration. Biol. Targets Ther. 2020, 14, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lommatzsch, A.; Hermans, P. Are low inflammatory reactions involved in exudative age-related macular degeneration? Graefe Arch. Clin. Exp. Ophthalmol. 2008, 246, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Peyman, G.A. Immunohistological study of subretinal membranes in age-related macular degeneration. Jpn. J. Ophthalmol. 1992, 36, 443–451. [Google Scholar] [PubMed]
- Nozaki, M.; Raisler, B.J. Drusen complement components c3a and c5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328. [Google Scholar] [CrossRef] [PubMed]
- Trakkides, T.-O.; Schäfer, N. Oxidative stress increases endogenous complement-dependent inflammatory and angiogenic responses in retinal pigment epithelial cells independently of exogenous complement sources. Antioxidants 2019, 8, 548. [Google Scholar] [CrossRef]
- Elsner, J.; Oppermann, M. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur. J. Immunol. 1994, 24, 518–522. [Google Scholar] [CrossRef]
- Liao, D.S.; Grossi, F.V. Complement c3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: A randomized phase 2 trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef]
- Focus: First in Human Study to Evaluate the Safety and Efficacy of gt005 Administered in Subjects with Dry amd. Available online: https://clinicaltrials.gov/ct2/show/NCT03846193 (accessed on 22 January 2021).
- Cornel, S.; Adriana, I.D. Anti-vascular endothelial growth factor indications in ocular disease. Rom. J. Ophthalmol. 2015, 59, 235–242. [Google Scholar]
- Pechan, P.; Wadsworth, S. Gene therapies for neovascular age-related macular degeneration. Cold Spring Harb. Perspect. Med. 2014, 5, a017335. [Google Scholar] [CrossRef]
- Honda, M.; Sakamoto, T. Experimental subretinal neovascularization is inhibited by adenovirus-mediated soluble vegf/flt-1 receptor gene transfection: A role of vegf and possible treatment for srn in age-related macular degeneration. Gene Ther. 2000, 7, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-M.; Estcourt, M. Preclinical safety evaluation of subretinal aav2. Sflt-1 in non-human primates. Gene Ther. 2012, 19, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.K.; Shen, W.Y. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene 2002, 9, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-M.; Shen, W.-Y. Long-term evaluation of aav-mediated sflt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol. Ther. 2005, 12, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-M.; Estcourt, M.J. rAAV. sFLT-1 gene therapy achieves lasting reversal of retinal neovascularization in the absence of a strong immune response to the viral vector. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4279–4287. [Google Scholar] [CrossRef]
- Maclachlan, T.K.; Lukason, M. Preclinical safety evaluation of aav2-sflt01- a gene therapy for age-related macular degeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 326–334. [Google Scholar] [CrossRef]
- The First Fda Approved Gene Therapy for Inherited Retinal Disease. Available online: https://www.ophth.wisc.edu/blog/2018/02/13/fda-approved-retinal-gene-therapy-luxturna-uw/ (accessed on 1 December 2020).
- Gene Therapy May Represent Future of Amd Treatment. Available online: https://www.healio.com/news/ophthalmology/20201020/gene-therapy-may-represent-future-of-amd-treatment (accessed on 1 December 2020).
- Heier, J.S.; Kherani, S. Intravitreous injection of aav2-sflt01 in patients with advanced neovascular age-related macular degeneration: A phase 1, open-label trial. Lancet 2017, 390, 50–61. [Google Scholar] [CrossRef]
- Bordet, T.; Behar-Cohen, F. Ocular gene therapies in clinical practice: Viral vectors and nonviral alternatives. Drug Discov. Today 2019, 24, 1685–1693. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Schachar, R.A. Phase 1 dose-escalation study of a sirna targeting the rtp801 gene in age-related macular degeneration patients. Eye 2012, 26, 1099–1105. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Classification | Clinical Manifestation |
|---|---|
| No AMD | No drusen and no RPE abnormalities |
| Normal aging changes | Drusen ≤ 63 μm and no RPE abnormalities |
| Early AMD | Drusen > 63 μm and ≤125 μm and no RPE abnormalities |
| Intermediate AMD | Drusen > 125 μm and/or RPE abnormalities |
| Late AMD | GA and/or neovascular AMD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22, 1296. https://doi.org/10.3390/ijms22031296
Ruan Y, Jiang S, Gericke A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. International Journal of Molecular Sciences. 2021; 22(3):1296. https://doi.org/10.3390/ijms22031296
Chicago/Turabian StyleRuan, Yue, Subao Jiang, and Adrian Gericke. 2021. "Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels" International Journal of Molecular Sciences 22, no. 3: 1296. https://doi.org/10.3390/ijms22031296
APA StyleRuan, Y., Jiang, S., & Gericke, A. (2021). Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. International Journal of Molecular Sciences, 22(3), 1296. https://doi.org/10.3390/ijms22031296