
Complete Androgen Insensitivity Syndrome: From Bench to Bed

 , , and
, , and 
Abstract
1. Introduction
2. Molecular Biology
3. Clinical Features
4. Oncological Risk
5. Hormonal Substitutive Therapy
6. Bone Health
7. HRT and Psychological Well-Being
8. Immune and Metabolic Aspects
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| AR | Androgen receptor gene |
| BMD | Bone mineral density |
| CAIS | Complete androgen insensitivity syndrome |
| HRT | Hormonal substitutive therapy |
References
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef] [PubMed]
- Galani, A.; Kitsiou-Tzeli, S.; Sofokleous, C.; Kanavakis, E.; Kalpini-Mavrou, A. Androgen insensitivity syndrome: Clinical features and molecular defects. Hormones 2008, 7, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Werner, R.; Bunch, T.; Hiort, O. Androgen insensitivity syndrome. Semin Reprod. Med. 2012, 30, 432–442. [Google Scholar] [CrossRef]
- Costagliola, G.; Cosci O di Coscio, M.; Masini, B.; Baldinotti, F.; Caligo, M.A.; Tyutyusheva, N.; Sessa, M.R.; Peroni, D.; Bertelloni, S. Disorders of sexual development with XY karyotype and female phenotype: Clinical findings and genetic background in a cohort from a single centre. J. Endocrinol. Investig. 2021, 44, 145–151. [Google Scholar] [CrossRef] [PubMed]
- De Paula, G.B.; Barros, B.A.; Carpini, S.; Tincani, B.J.; Mazzola, T.N.; Sanches Guaragna, M.; Piveta, C.S.; de Oliveira, L.C.; Andrade, J.G.; Guaragna-Filho, G.; et al. 408 cases of genital ambiguity followed by single multidisciplinary team during 23 years: Etiologic diagnosis and sex of rearing. Int. J. Endocrinol. 2016, 2016, 4963574. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.; Singla, M.; Vaiphei, K.; Kumar, S.; Bhansali, A. Disorders of sex development: A study of 194 cases. Endocr. Connect. 2018, 7, 364–371. [Google Scholar] [CrossRef]
- Berglund, A.; Johannsen, T.H.; Stochholm, K.; Viuff, M.H.; Fedder, J.; Main, K.M.; Gravholt, C.H. Incidence, prevalence, diagnostic delay, and clinical presentation of female 46,XY disorders of sex development. J. Clin. Endocrinol. Metab. 2016, 101, 4532–4540. [Google Scholar] [CrossRef]
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.S.; Gomes, N.L.; Faria, J.A., Jr.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; Mendonca, B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018, 62, 227–235. [Google Scholar] [CrossRef]
- Gulía, C.; Baldassarra, S.; Zangari, A.; Briganti, V.; Gigli, S.; Gaffi, M.; Signore, F.; Vallone, C.; Nucciotti, R.; Costantini, F.M.; et al. Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3873–3887. [Google Scholar]
- Gobinet, J.; Poujol, N.; Sultan, C. Molecular action of androgens. Mol. Cell. Endocrinol. 2002, 198, 15–24. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Hiort, O.; Sinnecker, G.H.; Holterhus, P.M.; Nitsche, E.M.; Kruse, K. Inherited and de novo androgen receptor gene mutations: Investigation of single-case families. J. Pediatr. 1998, 132, 939–943. [Google Scholar] [CrossRef]
- Achermann, J.C.; Hughes, I.A. Disorders of sexual development. In William Textbook of Endocrinology, 11th ed.; Kronenberg, H.M., Melmed, S., Polonsky, K.S., Larsen, P.R., Eds.; WB Saunders: Philadelphia, PA, USA, 2008; pp. 800–836. [Google Scholar]
- Audi, L.; Fernández-Cancio, M.; Carrascosa, A.; Andaluz, P.; Torán, N.; Piró, C.; Vilaró, E.; Vicens-Calvet, E.; Gussinyé, M.; Albisu, M.A.; et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J. Clin. Endocrinol. Metab. 2010, 95, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Deeb, A.; Hughes, I.A. Inguinal hernia in female infants: A cue to check the sex chromosomes? BJU Int. 2005, 96, 401–403. [Google Scholar] [CrossRef]
- Bertelloni, S. Gonadal surgery in complete androgen insensitivity syndrome: A debate. Sex Dev. 2017, 11, 169–170. [Google Scholar] [CrossRef]
- Chaudhry, S.; Tadokoro-Cuccaro, R.; Hannema, S.E.; Acerini, C.L.; Hughes, I.A. Frequency of gonadal tumors in complete androgen insensitivity syndrome (CAIS): A retrospective case-series analysis. J. Pediatr. Urol. 2017, 13, 498.e1–498.e6. [Google Scholar] [CrossRef]
- Tack, L.J.W.; Maris, E.; Looijenga, L.H.J.; Hannema, S.E.; Audi, L.; Köhler, B.; Holterhus, P.M.; Riedl, S.; Wisniewski, A.; Flück, C.E.; et al. Management of gonads in adults with androgen insensitivity: An international survey. Horm. Res. Paediatr. 2018, 90, 236–246. [Google Scholar] [CrossRef]
- Deans, R.; Creighton, S.M.; Liao, L.M.; Conway, G.S. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin. Endocrinol. 2012, 76, 894–898. [Google Scholar] [CrossRef]
- Döhnert, U.; Wünsch, L.; Hiort, O. Gonadectomy in complete androgen insensitivity syndrome: Why and when? Sex Dev. 2017, 11, 171–174. [Google Scholar] [CrossRef]
- Cools, M.; Looijenga, L. Update on the pathophysiology and risk factors for the development of malignant testicular germ cell tumors in complete androgen insensitivity syndrome. Sex Dev. 2017, 11, 175–181. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Kao, C.S.; Idrees, M.T. Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int. J. Mol. Sci. 2019, 20, 5017. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; Van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018, 23, 3392–3406. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Dati, E.; Baroncelli, G.I.; Hiort, O. Hormonal management of complete androgen insensitivity syndrome from adolescence onward. Horm. Res. Paediatr. 2011, 76, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, W.; Bertelloni, S. Sex hormone replacement in disorders of sex development. Endocr. Dev. 2014, 27, 149–159. [Google Scholar]
- Nordenström, A.; Röhle, R.; Thyen, U.; Bouvattier, C.; Slowikowska-Hilczer, J.; Reisch, N.; Claahsen van der Grinten, H.; Brac de la Perriere, A.; Cohen-Kettenis, P.T.; Köhler, B.; et al. Hormone therapy and patient satisfaction with treatment, in a large cohort of diverse disorders of sex development. Clin. Endocrinol. 2018, 88, 397–408. [Google Scholar] [CrossRef]
- Birnbaum, W.; Marshall, L.; Werner, R.; Kulle, A.; Holterhus, P.M.; Rall, K.; Köhler, B.; Richter-Unruh, A.; Hartmann, M.F.; Wudy, S.A.; et al. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: A multicentre, randomised, double-dummy, double-blind crossover trial. Lancet Diabetes Endocrinol. 2018, 6, 771–780. [Google Scholar] [CrossRef]
- Doehnert, U.; Bertelloni, S.; Werner, R.; Dati, E.; Hiort, O. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex Dev. 2015, 9, 69–74. [Google Scholar] [CrossRef]
- Baroncelli, G.I.; Bertelloni, S. The effects of sex steroids on bone growth. In Osteoprosisis in Men; Orwoll, E.S., Bilezikian, J.P., Vanderschueeren, D., Eds.; Academic Press: London, UK, 2010; pp. 105–118. [Google Scholar]
- Chen, J.F.; Lin, P.W.; Tsai, Y.R.; Yang, Y.C.; Kang, H.Y. Androgens and androgen receptor actions on bone health and disease: From androgen deficiency to androgen therapy. Cells 2019, 8, 1318. [Google Scholar] [CrossRef]
- Colvard, D.S.; Eriksen, E.F.; Keeting, P.E.; Wilson, E.M.; Lubahn, D.B.; French, F.S.; Riggs, B.L.; Spelsberg, T.C. Identification of androgen receptors in normal human osteoblast-like cells. Proc. Natl. Acad. Sci. USA 1989, 86, 854–857. [Google Scholar] [CrossRef]
- Iqbal, J.; Sun, L.; Kumar, T.R.; Blair, H.C.; Zaidi, M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc. Natl. Acad. Sci. USA 2006, 103, 14925–14930. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.; Lizneva, D.; Kim, S.M.; Sun, L.; Iqbal, J.; New, M.I.; Rosen, C.J.; Yuen, T. FSH, bone mass, body fat, and biological aging. Endocrinology 2018, 159, 3503–3514. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Peng, Y.; Sharrow, A.C.; Iqbal, J.; Zhang, Z.; Papachristou, D.J.; Zaidi, S.; Zhu, L.L.; Yaroslavskiy, B.B.; Zhou, H.; et al. FSH directly regulates bone mass. Cell 2006, 125, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Meriggiola, M.C.; Dati, E.; Balsamo, A.; Baroncelli, G.I. Bone mineral density in women living with complete androgen insensitivity syndrome and intact testes or removed gonads. Sex Dev. 2017, 11, 182–189. [Google Scholar] [CrossRef]
- Ji, Y.; Liu, P.; Yuen, T.; Haider, S.; He, J.; Romero, R.; Chen, H.; Bloch, M.; Kim, S.M.; Lizneva, D.; et al. Epitope-specific monoclonal antibodies to FSHβ increase bone mass. Proc. Natl. Acad. Sci. USA 2018, 115, 2192–2197. [Google Scholar] [CrossRef]
- Soule, S.G.; Conway, G.; Prelevic, G.M.; Prentice, M.; Ginsburg, J.; Jacobs, H.S. Osteopenia as a feature of the androgen insensitivity syndrome. Clin. Endocrinol. 1995, 43, 671–675. [Google Scholar] [CrossRef]
- Bertelloni, S.; Baroncelli, G.I.; Federico, G.; Cappa, M.; Lala, R.; Saggese, G. Altered bone mineral density in patients with complete androgen insensitivity syndrome. Horm. Res. 1998, 50, 309–314. [Google Scholar] [CrossRef]
- Marcus, R.; Leary, D.; Schneider, D.; Shane, E.; Favus, M.; Quigley, C.A. The contribution of testosterone to skeletal development and maintenance: Lessons from the androgen insensitivity syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 1032–1037. [Google Scholar] [CrossRef]
- Tian, Q.J.; Dai, Z.Q.; Yu, W.; Tian, J.P.; Lang, J.H. Study of bone mineral density in complete androgen insensitivity syndrome patients. Zhonghua Fu Chan Ke Za Zhi 2005, 40, 799–802. (In Chinese) [Google Scholar]
- Sobel, V.; Schwartz, B.; Zhu, Y.S.; Cordero, J.J.; Imperato-McGinley, J. Bone mineral density in the complete androgen insensitivity and 5a-reductase-2 deficiency syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 3017–3023. [Google Scholar] [CrossRef]
- Danilovic, D.L.; Correa, P.H.; Costa, E.M.; Melo, K.F.; Mendonca, B.B.; Arnhold, I.J. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos. Int. 2007, 18, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Goswami, D.; Trikudanathan, S.; Creighton, S.M.; Conway, G.S. Comparison of bone mineral density and body proportions between women with complete androgen insensitivity syndrome and women with gonadal dysgenesis. Eur. J. Endocrinol. 2008, 159, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gava, G.; Mancini, I.; Orsili, I.; Bertelloni, S.; Alvisi, S.; Seracchioli, R.; Meriggiola, M.C. Bone mineral density, body composition and metabolic profiles in adult women with complete androgen insensitivity syndrome and removed gonads using oral or transdermal estrogens. Eur. J. Endocrinol. 2019, 181, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Taes, Y.; Lapauw, B.; Vandewalle, S.; Zmierczak, H.; Goemaere, S.; Vanderschueren, D.; Kaufman, J.M.; T’Sjoen, G. Estrogen-specific action on bone geometry and volumetric bone density: Longitudinal observations in an adult with complete androgen insensitivity. Bone 2009, 45, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Minto, C.L.; Liao, K.L.; Conway, G.S.; Creighton, S.M. Sexual function in women with complete androgen insensitivity syndrome. Fertil. Steril. 2003, 80, 157–164. [Google Scholar] [CrossRef]
- Wu, M.V.; Manoli, D.S.; Fraser, E.J.; Coats, J.K.; Tollkuhn, J.; Honda, S.; Harada, N.; Shah, N.M. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 2009, 139, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Slob, A.K.; van der Werff ten Bosch, J.J.; van Hall, E.V.; de Jong, F.H.; Weijmar Schultz, W.C.; Eikelboom, F.A. Psychosexual functioning in women with complete testicular feminization: Is androgen replacement therapy preferable to estrogen? J. Sex Marital Ther. 1993, 19, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Yeh, S.; Lee, S.O.; Chang, T.M. Androgen receptor (AR) pathophysiological roles in androgen-related diseases in skin, bone/muscle, metabolic syndrome and neuron/immune systems: Lessons learned from mice lacking AR in specific cells. Nucl. Recept. Signal. 2013, 11, e001. [Google Scholar] [CrossRef]
- Chuang, K.H.; Altuwaijri, S.; Li, G.; Lai, J.J.; Chu, C.Y.; Lai, K.P.; Lin, H.Y.; Hsu, J.W.; Keng, P.; Wu, M.C.; et al. Neutropenia with impaired host defense against microbial infection in mice lacking androgen receptor. J. Exp. Med. 2009, 206, 1181–1199. [Google Scholar] [CrossRef]
- Lin, H.Y.; Xu, Q.; Yeh, S.; Wang, R.S.; Sparks, J.D.; Chang, C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes 2005, 54, 1717–1725. [Google Scholar] [CrossRef]
- Dati, E.; Baroncelli, G.I.; Mora, S.; Russo, G.; Baldinotti, F.; Parrini, D.; Erba, P.; Simi, P.; Bertelloni, S. Body composition and metabolic profile in women with complete androgen insensitivity syndrome. Sex Dev. 2009, 3, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Nordenström, A.; Robeva, R.; Hall, J.; Westerveld, P.; Flück, C.; Köhler, B.; Berra, M.; Springer, A.; Schweizer, K.; et al. COST Action BM1303 working group 1. Caring for individuals with a difference of sex development (DSD): A Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 415–429. [Google Scholar] [CrossRef] [PubMed]



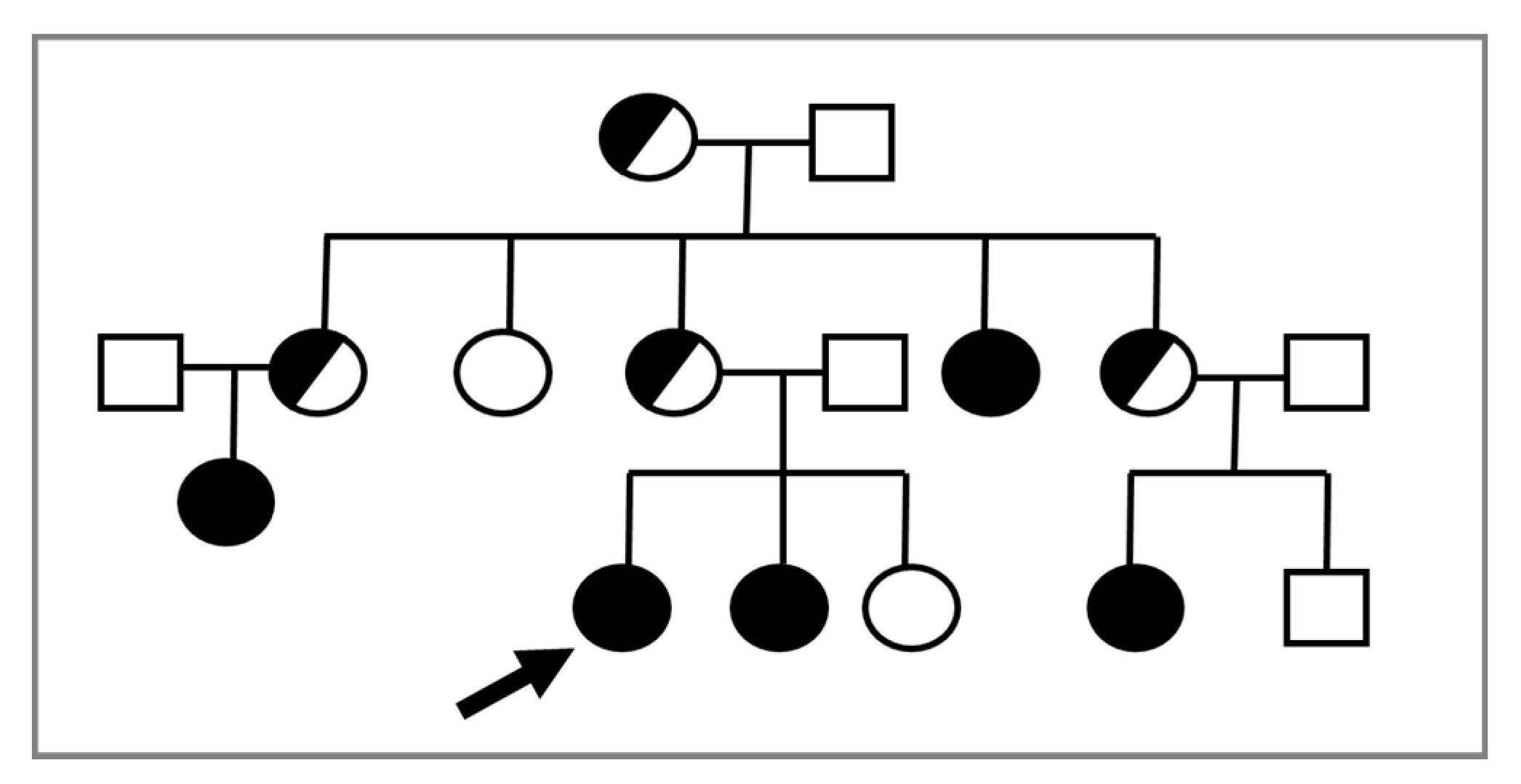{kind=link}
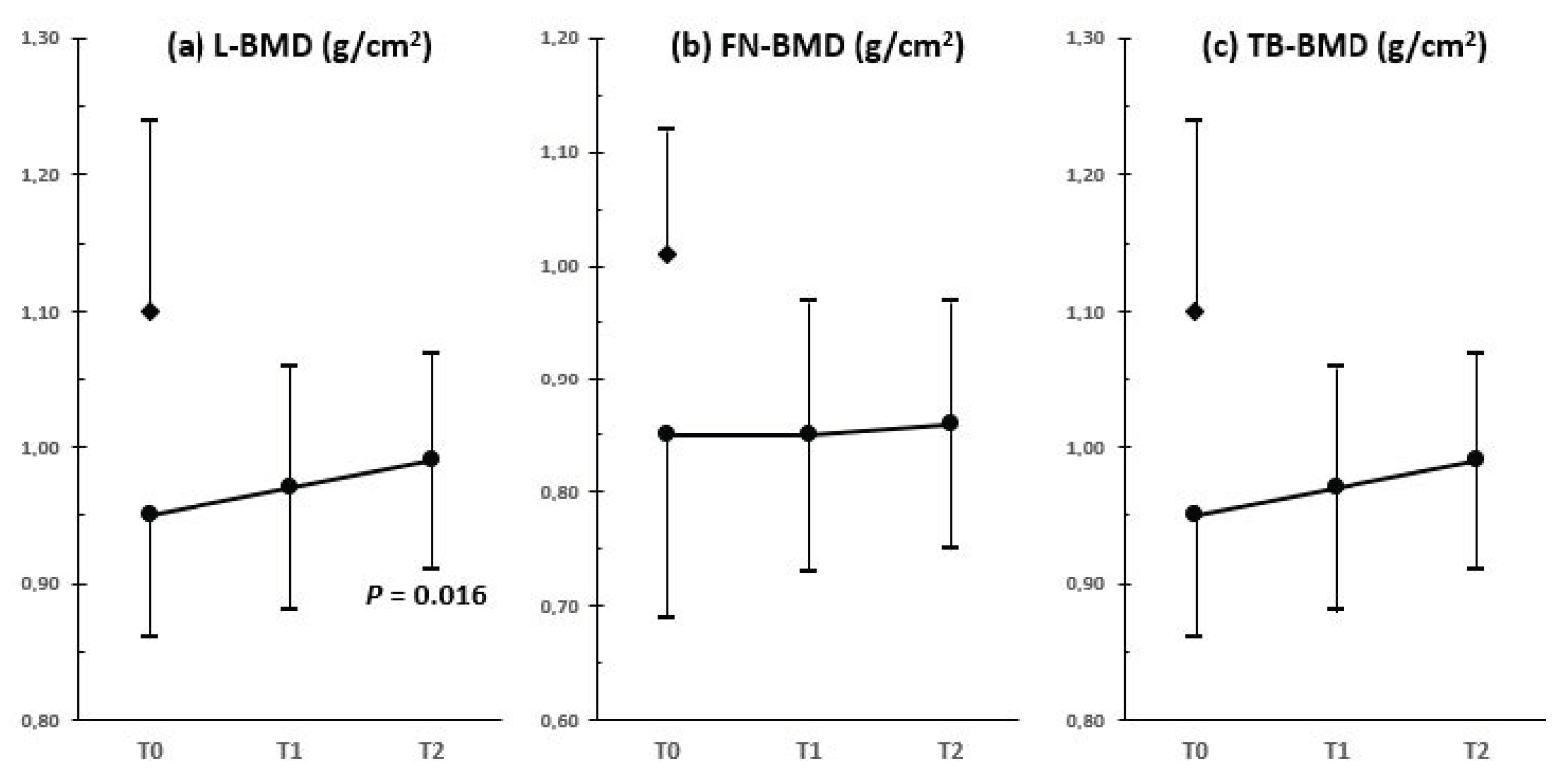{kind=link}
| Type of Genetic Variants | Pisa a | McGill b | HGMD c |
|---|---|---|---|
| Point missense or nonsense mutations | 65.1% | 50.0% | 75.0% |
| Insertions or deletions | 12.1% | 28.0% | 6.6% |
| Intronic or intron–exon junction point mutations | 12.15% | 5.4% | 4.0% |
| Small or gross deletions of the AR gene | 7.6% | 6.7% | 14.4% |
| Complete deletions of the AR gene | 3.0% | 1.3% | ¾ |
| Factor(s) | Mechanism(s) |
|---|---|
| Complete AR resistance | Abnormal activity of osteoblasts and osteocytes. |
| Iatrogenic hypergonadotropic hypogonadism | Inadequate hormonal substitutive therapy. |
| Absence of testicular INSL-3 (reduced bone mass through dysregulation of osteoblastic differentiation, deposition of the bone matrix and osteoclastogenesis; altered functioning of the musculo-skeletal unit in postnatal life). | |
| High FSH levels (greater bone resorption by osteoclasts due to the activation of the FSH receptor *) | |
| Others | Age at gonadectomy (before or after achievement of peak bone mass). |
| Type, doses, compliance and duration of HRT. | |
| Unhealthy eating, low calcium intake, reduced physical activity, inadequate muscle mass, smoking and low vitamin D. | |
| Aging. |
| Author | n | Age | Genetic Analysis | HRT Compliance | BMD | ||
|---|---|---|---|---|---|---|---|
| Lumbar | Femoral | R.V. | |||||
| Soule et al. [38] | 6 | 13–38 | no | v.c. a | ↓↓ | ↓↓ | F |
| Bertelloni et al. [39] | 10 | 4–20 | yes | b.g./v.c. | ↓↓ b | ND | F/M |
| Marcus et al. [40] | 22 | 11–65 | no | v.c. | ↓ d | N d | F |
| Tian et al. [41] | 14 | ↓↓ | ↓↓ | F/M | |||
| Sobel et al. [42] | 12 | 17–62 | yes/no | b.g./v.c. | ↓↓ c | ↓ c | F/M |
| Danilovic et al. [43] | 5 | 20–25 | yes | good/fair | ↓ b | N b | F/M |
| Han et al. [44] | 46 | 32.2 | yes/no | good | ↓↓ | ↓ | F |
| Gava et al. [45] | 32 | 19–57 | yes | good | ↓ | ↓ | F |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyutyusheva, N.; Mancini, I.; Baroncelli, G.I.; D’Elios, S.; Peroni, D.; Meriggiola, M.C.; Bertelloni, S. Complete Androgen Insensitivity Syndrome: From Bench to Bed. Int. J. Mol. Sci. 2021, 22, 1264. https://doi.org/10.3390/ijms22031264
Tyutyusheva N, Mancini I, Baroncelli GI, D’Elios S, Peroni D, Meriggiola MC, Bertelloni S. Complete Androgen Insensitivity Syndrome: From Bench to Bed. International Journal of Molecular Sciences. 2021; 22(3):1264. https://doi.org/10.3390/ijms22031264
Chicago/Turabian StyleTyutyusheva, Nina, Ilaria Mancini, Giampiero Igli Baroncelli, Sofia D’Elios, Diego Peroni, Maria Cristina Meriggiola, and Silvano Bertelloni. 2021. "Complete Androgen Insensitivity Syndrome: From Bench to Bed" International Journal of Molecular Sciences 22, no. 3: 1264. https://doi.org/10.3390/ijms22031264
APA StyleTyutyusheva, N., Mancini, I., Baroncelli, G. I., D’Elios, S., Peroni, D., Meriggiola, M. C., & Bertelloni, S. (2021). Complete Androgen Insensitivity Syndrome: From Bench to Bed. International Journal of Molecular Sciences, 22(3), 1264. https://doi.org/10.3390/ijms22031264