Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Innate Immune System of the Brain
3. Cellular Sentinels of the Brain
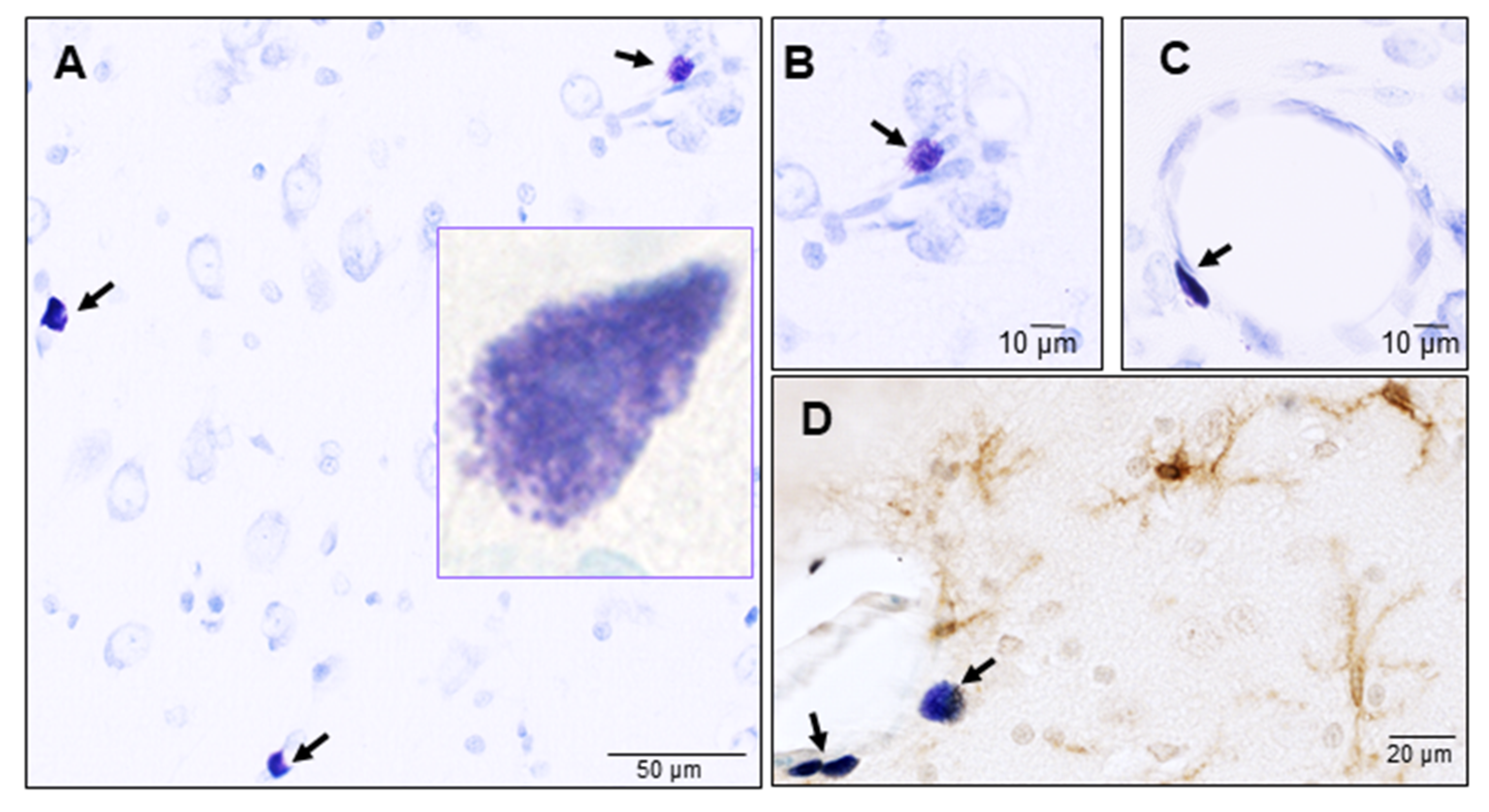
3.1. Mast Cells
Mast Cells: Guardians of the Gateway to the Brain
3.2. Microglia
3.2.1. Homeostatic Microglia: Nurturers of the Brain
3.2.2. Homeostatic Microglia: Sentinels of the Brain
4. Mast Cell Activation in Neurodegeneration
4.1. Mast Cells: Guardians of Homeostasis in the Brain
4.2. Mast Cells: Warriors of the Brain
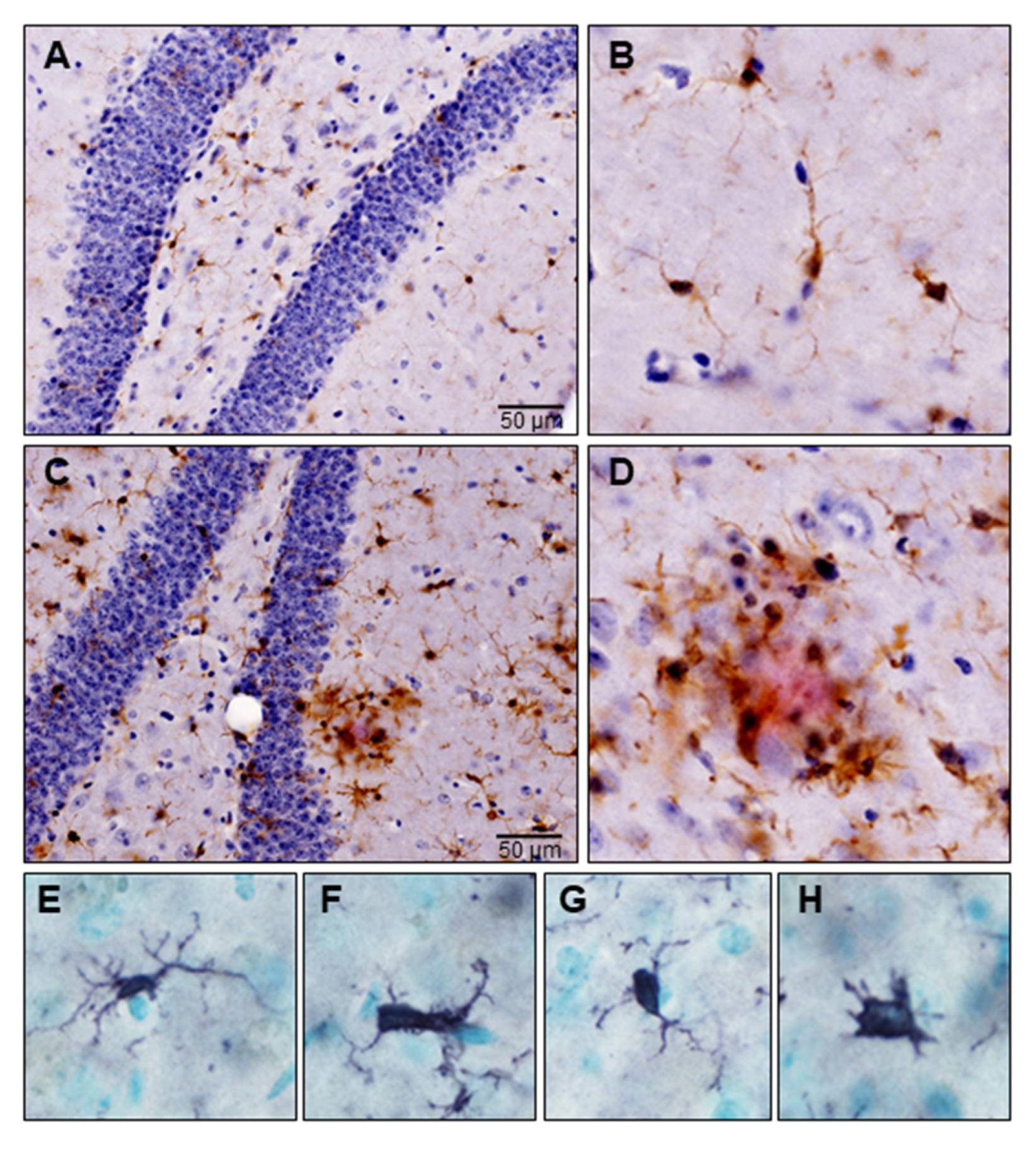
5. Microglial Activation in Neurodegeneration
5.1. Disease-Associated Microglia: Sentinels of the Brain
5.2. Disease-Associated Microglia: Warriors of the Brain
5.3. Disease-Associated Microglia: Enemies within the Brain
6. Mast Cell-Microglia Interactions in Neurodegeneration
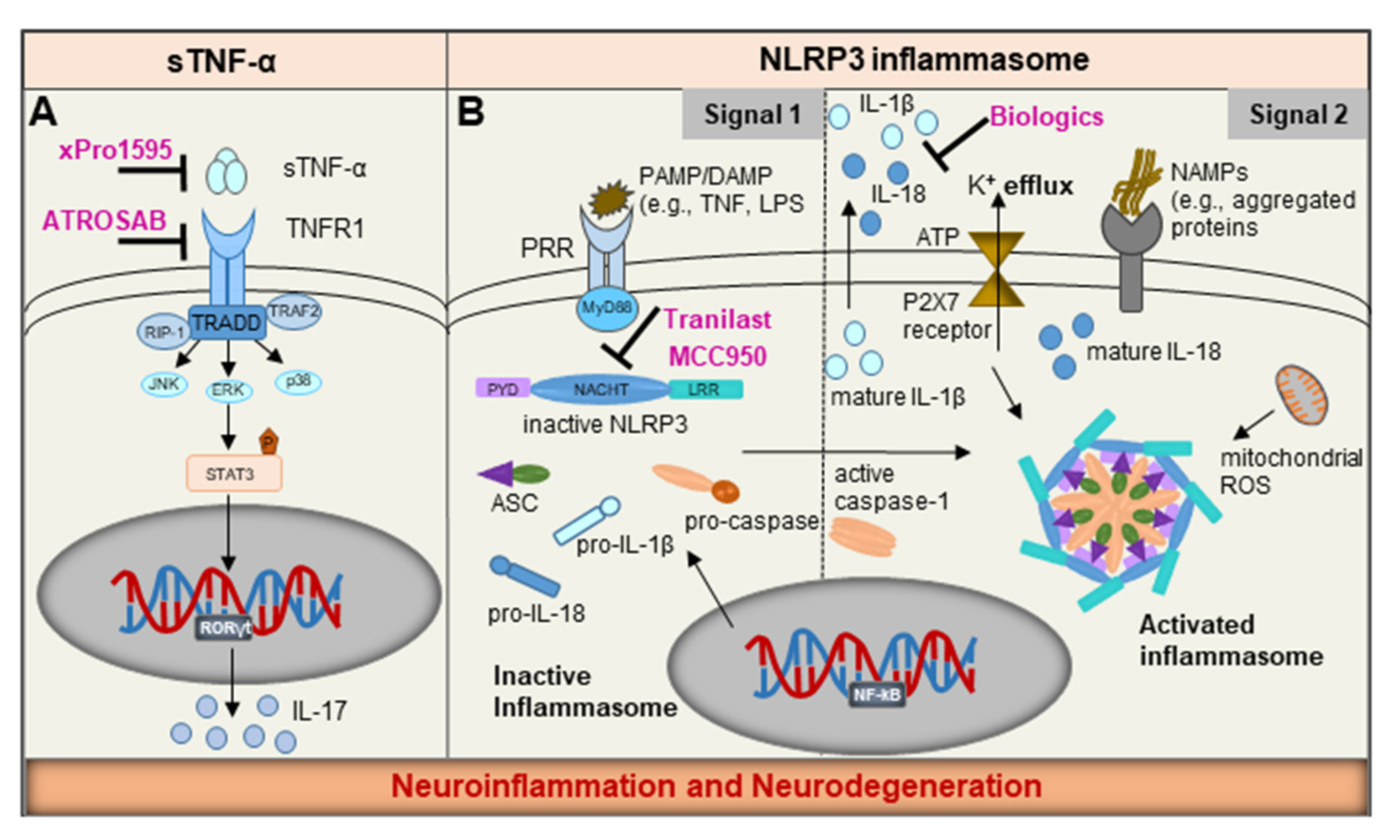
NLRP3 Inflammasome: Common Sensor in Microglia and Mast Cells
7. Emerging Therapeutics
7.1. NLRP3 Inflammasome—A ‘Druggable’ Target
7.2. Precision Targeting of Soluble TNF-α
8. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Emerging Concepts in Alzheimer’s Disease. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 291–319. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Jetté, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El, K.J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 2017, 23, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, E.; Van Bergeijk, D.; Oosting, R.S.; Redegeld, F. Mast cells in neuroinflammation and brain disorders. Neurosci. Biobehav. Rev. 2017, 79, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Nair, A.; Gupta, M. Mast Cells in Neurodegenerative Disease. Front. Cell. Neurosci. 2019, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and mast cells: Two tracks on the road to neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2016, 101, 87–98. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; De Oliveira, A.C. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef]
- McKee, C.M.; Coll, R.C. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 2020, 108, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: An immune sensor of cellular stress and infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795. [Google Scholar] [CrossRef] [PubMed]
- De, N.D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Beaven, M.A. Our perception of the mast cell from Paul Ehrlich to now. Eur. J. Immunol. 2009, 39, 11–25. [Google Scholar] [CrossRef]
- Okayama, Y.; Kawakami, T. Development, Migration, and Survival of Mast Cells. Immunol. Res. 2006, 34, 97–116. [Google Scholar] [CrossRef]
- Janssens, A.S.; Heide, R.; Den Hollander, J.C.; Mulder, P.G.; Tank, B.; Oranje, A.P. Mast cell distribution in normal adult skin. J. Clin. Pathol. 2005, 58, 285–289. [Google Scholar] [CrossRef]
- Silver, R.; Silverman, A.-J.; Vitković, L.; Lederhendler, I. Mast cells in the brain: Evidence and functional significance. Trends Neurosci. 1996, 19, 25–31. [Google Scholar] [CrossRef]
- Florenzano, F.; Bentivoglio, M. Degranulation, density, and distribution of mast cells in the rat thalamus: A light and electron microscopic study in basal conditions and after intracerebroventricular administration of nerve growth factor. J. Comput. Neurol. 2000, 424, 651–669. [Google Scholar] [CrossRef]
- Khalil, M.; Ronda, J.; Weintraub, M.; Jain, K.; Silver, R.; Silverman, A.J. Brain mast cell relationship to neurovasculature during development. Brain Res. 2007, 1171, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Macchi, V.; Parenti, A.; De Caro, R. The distribution of mast cells in the human area postrema. J. Anat. 2004, 204, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hough, L.B. Cellular localization and possible functions for brain histamine: Recent progress. Prog. Neurobiol. 1988, 30, 469–505. [Google Scholar] [CrossRef]
- Silverman, A.-J.; Sutherland, A.K.; Wilhelm, M.; Silver, R. Mast Cells Migrate from Blood to Brain. J. Neurosci. 2000, 20, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.B.; Siman, R.; Iqbal, M.A.; Potter, H. Identification of a chymotrypsin-like mast cell protease in rat brain capable of generating the N-terminus of the Alzheimer amyloid beta-protein. J. Neurochem. 1993, 61, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Shanas, U.; Bhasin, R.; Sutherland, A.K.; Silverman, A.-J.; Silver, R. Brain mast cells lack the c-kit receptor: Immunocytochemical evidence. J. Neuroimmunol. 1998, 90, 207–211. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Pang, X.; Letourneau, R.; Rozniecki, J.; Wang, L.; Theoharides, T. Definitive characterization of rat hypothalamic mast cells. Neuroscience 1996, 73, 889–902. [Google Scholar] [CrossRef]
- Zhang, B.; Asadi, S.; Weng, Z.; Sismanopoulos, N.; Theoharides, T.C. Stimulated Human Mast Cells Secrete Mitochondrial Components That Have Autocrine and Paracrine Inflammatory Actions. PLoS ONE 2012, 7, e49767. [Google Scholar] [CrossRef]
- Beghdadi, W.; Madjène, L.C.; Benhamou, M.; Charles, N.; Gautier, G.; Launay, P.; Blank, U. Mast cells as cellular sensors in inflammation and immunity. Front. Immunol. 2011, 2, 37. [Google Scholar] [CrossRef]
- John, A.L.S.; Abraham, S.N. Innate Immunity and Its Regulation by Mast Cells. J. Immunol. 2013, 190, 4458–4463. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; King, B.; Silverman, A.J.; Silver, R. Gonadal steroids regulate the number and activational state of mast cells in the medial habenula. Endocrinology 2000, 141, 1178–1186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boyce, J.A. Mast cells and eicosanoid mediators: A system of reciprocal paracrine and autocrine regulation. Immunol. Rev. 2007, 217, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Lu, H.; Miranda, A.S.; Ransohoff, R.M. Ontogeny and Functions of Central Nervous System Macrophages. J. Immunol. 2014, 193, 2615–2621. [Google Scholar] [CrossRef]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, P.; Attwell, D.; Madry, C. Ion Channels and Receptors as Determinants of Microglial Function. Trends Neurosci. 2019, 42, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, P.E.; Liberatore, G.T.; Wong, J.Y.F.; Porritt, M.J.; Frerichs, F.; Donnan, G.A.; Howells, D.W. Activated Macrophages and Microglia Induce Dopaminergic Sprouting in the Injured Striatum and Express Brain-Derived Neurotrophic Factor and Glial Cell Line-Derived Neurotrophic Factor. J. Neurosci. 1999, 19, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Stevens, B. Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef]
- Dringen, R. Oxidative and Antioxidative Potential of Brain Microglial Cells. Antioxid. Redox Signal. 2005, 7, 1223–1233. [Google Scholar] [CrossRef]
- Cartier, L.; Hartley, O.; Dubois-Dauphin, M.; Krause, K.-H. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res. Rev. 2005, 48, 16–42. [Google Scholar] [CrossRef]
- Thompson, K.K.; Tsirka, S.E. The Diverse Roles of Microglia in the Neurodegenerative Aspects of Central Nervous System (CNS) Autoimmunity. Int. J. Mol. Sci. 2017, 18, 504. [Google Scholar] [CrossRef]
- Pratt, B.M.; McPherson, J.M. TGF-beta in the central nervous system: Potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor Rev. 1997, 8, 267–292. [Google Scholar] [CrossRef]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Hu, W.-W.; Wang, X.-F.; Yang, Y.; Lou, G.-D.; Jin, M.-M.; Yan, H.-J.; Zeng, W.-Z.; Shen, Y.; Zhang, S.-H.; et al. Histamine up-regulates astrocytic glutamate transporter 1 and protects neurons against ischemic injury. Neuropharmacology 2014, 77, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, S.; Lemmens, E.; Geurts, N.; Kramer, P.; Maurer, M.; Hendriks, J.J.; Hendrix, S. The role of mast cells in neuroinflammation. Acta Neuropathol. 2013, 125, 637–650. [Google Scholar] [CrossRef]
- Heldmann, U.; Thored, P.; Claaßen, J.; Arvidsson, A.; Kokaia, Z.; Lindvall, O. TNF-α antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp. Neurol. 2005, 196, 204–208. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.H.; McDermott, M.F. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]
- Mattila, O.S.; Strbian, D.; Saksi, J.; Pikkarainen, T.O.; Rantanen, V.; Tatlisumak, T.; Lindsberg, P.J. Cerebral Mast Cells Mediate Blood-Brain Barrier Disruption in Acute Experimental Ischemic Stroke Through Perivascular Gelatinase Activation. Stroke 2011, 42, 3600–3605. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Dong, H.; Xu, Y.; Zhang, S. Induction of Microglial Activation by Mediators Released from Mast Cells. Cell. Physiol. Biochem. 2016, 38, 1520–1531. [Google Scholar] [CrossRef]
- Maslinska, D.; Laure-Kamionowska, M.; Maslinski, K.T.; Gujski, M.; Maslinski, S. Distribution of tryptase-containing mast cells and metallothionein reactive astrocytes in human brains with amyloid deposits. Inflamm. Res. 2007, 56, S17–S18. [Google Scholar] [CrossRef]
- Harcha, P.A.; Vargas, A.; Yi, C.; Koulakoff, A.A.; Giaume, C.; Saez, J.C. Hemichannels Are Required for Amyloid be-ta-Peptide-Induced Degranulation and Are Activated in Brain Mast Cells of APPswe/PS1dE9 Mice. J. Neurosci. 2015, 35, 9526–9538. [Google Scholar] [CrossRef][Green Version]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a Potent and Selective Tyrosine Kinase Inhibitor Targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedrós, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Fattal, R.; Pattani, S.; Yang, E.; Zaheer, S.; Santillan, D.A.; Santillan, M.; Zaheer, A. Mast Cells Release Chemokine CCL2 in Response to Parkinsonian Toxin 1-Methyl-4-Phenyl-Pyridinium (MPP+). Neurochem. Res. 2015, 41, 1042–1049. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Kumar, K.K.; Yelam, A.; Kaur, H.; Dubova, I.; Raikwar, S.P.; et al. Glia Maturation Factor and Mast Cell-Dependent Expression of In-flammatory Mediators and Proteinase Activated Receptor-2 in Neuroinflammation. J. Alzheimer’s Dis. 2018, 66, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.U.; Cho, J.W.; Kim, S.Y.; Shin, J.H.; Ro, J.Y. Inflammatory mediators resulting from transglutaminase 2 expressed in mast cells contribute to the development of Parkinson’s disease in a mouse model. Toxicol. Appl. Pharmacol. 2018, 358, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progres-sion. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alz-heimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mech-anism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Emerit, J.; Edeas, M.A.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Pandey, S.; Ribecco-Lutkiewicz, M.; Monette, R.; Borowy-Borowski, H.; Walker, P.R.; Sikorska, M. Molecular mechanisms of glutamate neurotoxicity in mixed cultures of NT2-derived neurons and astrocytes: Protective effects of coenzyme Q10. J. Neurosci. Res. 2003, 72, 691–703. [Google Scholar] [CrossRef]
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Gupta, S.; Parrino, T.E.; Knight, A.G.; Ebenezer, P.J.; Weidner, A.M.; Levine, H.; Keller, J.N.; Markesbery, W.R. NOX Activity Is Increased in Mild Cognitive Impairment. Antioxid. Redox Signal. 2010, 12, 1371–1382. [Google Scholar] [CrossRef]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef]
- Lue, L.-F.; Rydel, R.; Brigham, E.F.; Yang, L.-B.; Hampel, H.; Murphy, G.M.; Brachova, L.; Yan, S.-D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Prà, I.; Rossi, F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hald, A.; Lotharius, J. Oxidative stress and inflammation in Parkinson’s disease: Is there a causal link? Exp. Neurol. 2005, 193, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Mucke, L. Inflammation in Neurodegenerative Disease—A Double-Edged Sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.-C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Brown, G.C.; St George-Hyslop, P.H. Deciphering microglial diversity in Alzheimer’s disease. Science 2017, 356, 1123–1124. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nat. Cell Biol. 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Galli, S.J.; Grimbaldeston, M.A.; Tsai, M. Immunomodulatory mast cells: Negative, as well as positive, regulators of immunity. Nat. Rev. Immunol. 2008, 8, 478–486. [Google Scholar] [CrossRef]
- Frick, L.; Rapanelli, M.; Abbasi, E.; Ohtsu, H.; Pittenger, C. Histamine regulation of microglia: Gene-environment interaction in the regulation of central nervous system inflammation. Brain Behav. Immun. 2016, 57, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Santos, T.; Gonçalves, J.; Baltazar, G.; Ferreira, L.; Agasse, F.; Bernardino, L. Histamine modulates microglia function. J. Neuroinflamm. 2012, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, W.; Zeng, X.; Hu, G.; Zhang, H.; He, S.-H.; Zhang, S. Histamine Induces Upregulated Expression of Histamine Receptors and Increases Release of Inflammatory Mediators from Microglia. Mol. Neurobiol. 2014, 49, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Barata-Antunes, S.; Cristovao, A.C.; Pires, J.; Rocha, S.M.; Bernardino, L. Dual role of histamine on microglia-induced neu-rodegeneration. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Panula, P.; Nuutinen, S. The histaminergic network in the brain: Basic organization and role in disease. Nat. Rev. Neurosci. 2013, 14, 472–487. [Google Scholar] [CrossRef]
- Rocha, S.M.; Saraiva, T.; Cristóvão, A.C.; Ferreira, R.; Santos, T.; Esteves, M.; Saraiva, C.; Je, G.; Cortes, L.; Valero, J.; et al. Histamine induces microglia activation and dopaminergic neuronal toxicity via H1 receptor activation. J. Neuroinflamm. 2016, 13, 1–16. [Google Scholar] [CrossRef]
- Yuan, H.; Zhu, X.; Zhou, S.; Chen, Q.; Zhu, X.; Ma, X.; He, X.; Tian, M.; Shi, X. Role of mast cell activation in inducing microglial cells to release neurotrophin. J. Neurosci. Res. 2009, 88, 1348–1354. [Google Scholar] [CrossRef]
- Wareham, K.; Vial, C.; Wykes, R.C.; Bradding, P.; Seward, E.P. Functional evidence for the expression of P2X1, P2X4 and P2X7 receptors in human lung mast cells. Br. J. Pharmacol. 2009, 157, 1215–1224. [Google Scholar] [CrossRef]
- Kempuraj, D.; Selvakumar, G.P.; Zaheer, S.; Thangavel, R.; Ahmed, M.E.; Raikwar, S.P.; Govindarajan, R.; Iyer, S.; Zaheer, A. Cross-Talk between Glia, Neurons and Mast Cells in Neuroinflammation Associated with Parkinson’s Disease. J. Neuroimmune Pharmacol. 2017, 13, 100–112. [Google Scholar] [CrossRef]
- Kempuraj, D.; Thangavel, R.; Selvakumar, G.P.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Zahoor, H.; Saeed, D.; Dubova, I.; Giler, G.; et al. Mast Cell Proteases Activate Astrocytes and Glia-Neurons and Release Interleukin-33 by Activating p38 and ERK1/2 MAPKs and NF-kappaB. Mol. Neurobiol. 2019, 56, 1681–1693. [Google Scholar] [CrossRef]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 Is Associated with a Faster Rate of Cognitive Decline during Early Stages of Alzheimer’s Disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Yamamoto, M.; Xiong, H.; Lambert, M.P.; Klein, W.L.; Gendelman, H.E.; Ransohoff, R.M.; Ikezu, T. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS ONE 2009, 4, e6197. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Lin, S.; He, L.; Zhu, X.; Zhou, Z.; Chen, S.; Wang, Y.; Ding, J. Association of Two Polymorphisms in CCL2 with Parkinson’s Disease: A Case-Control Study. Front Neurol. 2019, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reac-tions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Bonnekoh, H.; Scheffel, J.; Kambe, N.; Krause, K. The role of mast cells in autoinflammation. Immunol. Rev. 2018, 282, 265–275. [Google Scholar] [CrossRef]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple Binding Sites on the Pyrin Domain of ASC Protein Allow Self-association and Interaction with NLRP3 Protein*. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Nambayan, R.J.T.; Sandin, S.I.; Quint, D.A.; Satyadi, D.M.; De Alba, E. The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains, PYD and CARD. J. Biol. Chem. 2019, 294, 439–452. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Heneka, M.T. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019, 33, 13075–13084. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De, N.D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate in-flammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amy-loid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio, A.A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.; Cooper, M.A.; O’Neill, L.A.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopa-minergic neurodegeneration in mice. Sci. Trans. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Park. Dis. 2018, 4, 1–9. [Google Scholar] [CrossRef]
- Chatterjee, K.; Roy, A.; Banerjee, R.; Choudhury, S.; Mondal, B.; Halder, S.; Basu, P.; Shubham, S.; Dey, S.; Kumar, H. Inflammasome and α-synuclein in Parkinson’s disease: A cross-sectional study. J. Neuroimmunol. 2019, 338, 577089. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pan, Y.-T.; Zhang, Z.-Y.; Yang, H.; Yu, S.-Y.; Zheng, Y.; Ma, J.-H.; Wang, X.-M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopa-minergic neurodegeneration. Cell Death. Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kambe, N.; Saito, M.; Nishikomori, R.; Kim, Y.G.; Murakami, M.; Núñez, G.; Matsue, H. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J. Exp. Med. 2009, 206, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Russi, A.E.; Walker-Caulfield, M.E.; Brown, M.A. Mast cell inflammasome activity in the meninges regulates EAE disease severity. Clin. Immunol. 2018, 189, 14–22. [Google Scholar] [CrossRef]
- Braddock, M.; Quinn, A. Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004, 3, 330–340. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targetsNLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Yang, W.; Sabi-Mouka, E.M.B.; Wang, L.; Shu, C.; Wang, Y.; Ding, J.; Ding, L. Determination of tranilast in bio-samples by LC–MS/MS: Application to a pharmacokinetic and brain tissue distribution study in rats. J. Pharm. Biomed. Anal. 2018, 147, 479–484. [Google Scholar] [CrossRef]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A com-prehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, W.; Magnus, T.; Martin, B.; Keselman, A.; Mattson, M.P.; Maudsley, S. Targeting TNF-alpha receptors for neuro-therapeutics. Trends Neurosci. 2008, 31, 504–511. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking Soluble Tumor Necrosis Factor Signaling with Dominant-Negative Tumor Necrosis Factor Inhibitor Attenuates Loss of Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; DesJarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF Signaling by Rationally Designed Dominant-Negative TNF Variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro(R)1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Parkinsons. Dis. 2014, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef]
- Cavanagh, C.; Tse, Y.C.; Nguyen, H.-B.; Krantic, S.; Breitner, J.C.; Quirion, R.; Wong, T.P. Inhibiting tumor necrosis factor-α before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 2016, 47, 41–49. [Google Scholar] [CrossRef]
- Zettlitz, K.A.; Lorenz, V.; Landauer, K.; Münkel, S.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs 2010, 2, 639–647. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and temporal heterogeneity of mouse and human microglia at sin-gle-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. https://doi.org/10.3390/ijms22031093
Sandhu JK, Kulka M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(3):1093. https://doi.org/10.3390/ijms22031093
Chicago/Turabian StyleSandhu, Jagdeep K., and Marianna Kulka. 2021. "Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 3: 1093. https://doi.org/10.3390/ijms22031093
APA StyleSandhu, J. K., & Kulka, M. (2021). Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(3), 1093. https://doi.org/10.3390/ijms22031093