Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus


Abstract
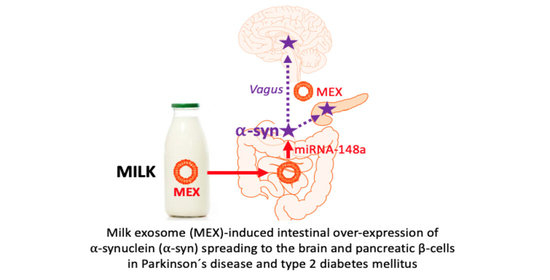
1. Introduction
2. Epidemiological Evidence
2.1. Milk Intake and Parkinson’s Disease
2.2. Milk Consumption and Type 2 Diabetes Mellitus
3. Milk: A Signaling System for Postnatal Growth and Differentiation
3.1. Milk Activates mTORC1 and Inhibits Autophagy
3.2. Milk Exosomes: Epigenetic Drivers of Developmental Genes
3.3. Milk Exosomes: Potential Drivers of α-Synuclein Expression and Transmission
3.4. Milk Exosomal miRNAs and SNCA Promoter Demethylation
4. Autophagy-Lysosome Pathway Controls Exosomal α-Synuclein Export
4.1. Milk Exosomes: Potential Modifiers of Chaperone-Mediated Autophagy
4.2. Bacterial Fermentation Degrades Milk Exosomes
5. Milk and Oxidative Stress
5.1. Galactose: Inducer of Oxidative Stress
5.2. Galactose: A Mitochondrial Stressor
5.3. Galactose: Inducer of miRNA-21
5.4. Oxidative Stress and Abelson Tyrosine Kinase Activation
5.5. Bacterial Fermentation Reduces Galactose Content of Dairy Products
5.6. Branched-Chain Amino Acids and Oxidative Stress
5.7. MiRNA-148a: Inhibitor of Mitochondrial Function and Autophagy
6. The α-Synuclein Link between Parkinson’s Disease and Type 2 Diabetes
6.1. α-Synuclein: Functional Component of β-Cells
6.2. Galactose Disturbs β-Cell α-Synuclein Homeostasis
7. Conclusions and Perspectives
8. Materials and Methods
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The incidence of Parkinson’s disease: A systematic review and meta-analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An Essay on the Shaking Palsy; Whittingham and Rowland for Sherwood, Needly and Jones: London, UK, 1817. [Google Scholar]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. Schol. Ed. 2014, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 1. [Google Scholar]
- Reich, S.G.; Savitt, J.M. Parkinson’s disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef]
- Campdelacreu, J. Parkinson disease and Alzheimer disease: Environmental risk factors. Neurologia 2014, 29, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P. Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Delamarre, A.; Meissner, W.G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse Med. 2017, 46 Pt 1, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Doneddu, P.E.; Riva, N.; Nobile-Orazio, E.; Quattrini, A. Diet, microbiota and brain health: Unraveling the network intersecting metabolism and neurodegeneration. Int. J. Mol. Sci. 2020, 21, 7471. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, S.; Litvan, I. Environmental exposures and Parkinson’s disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease—The gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef]
- Liddle, R.A. Parkinson’s disease from the gut. Brain Res. 2018, 1693, 201–206. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Bohórquez, D.V.; Liddle, R.A. Axon-like basal processes in enteroendocrine cells: Characteristics and potential targets. Clin. Transl. Sci. 2011, 4, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Bohórquez, D.V.; Samsa, L.A.; Roholt, A.; Medicetty, S.; Chandra, R. An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PLoS ONE 2014, 9, e899881. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Sternini, C.; De Giorgio, R.; Greenwood-Van Meerveld, B. Enteroendocrine cells: A review of their role in brain-gut communication. Neurogastroenterol. Motil. 2016, 28, 620–630. [Google Scholar] [CrossRef]
- Borghammer, P. How does Parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. 2018, 33, 48–57. [Google Scholar] [CrossRef]
- Steiner, J.A.; Quansah, E.; Brundin, P. The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res. 2018, 373, 161–173. [Google Scholar] [CrossRef]
- Chandra, R.; Hiniker, A.; Kuo, Y.M.; Nussbaum, R.L.; Liddle, R.A. α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2017, 2, e92295. [Google Scholar] [CrossRef]
- Bohórquez, D.V.; Liddle, R.A. The gut connectome: Making sense of what you eat. J. Clin. Investig. 2015, 125, 888–890. [Google Scholar] [CrossRef]
- Bohórquez, D.V.; Shahid, R.A.; Erdmann, A.; Kreger, A.M.; Wang, Y.; Calakos, N.; Wang, F.; Liddle, R.A. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Investig. 2015, 125, 782–786. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.B.; O’Callaghan, J.P. Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metabolism 2008, 57 (Suppl. 2), S44–S49. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Li, H.; Yan, H.; Zhang, P.; Chang, L.; Li, T. Risk of Parkinson disease in diabetes mellitus: An updated meta-analysis of population-based cohort studies. Medicine 2016, 95, e3549. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Renaud, J.; Simola, N.; Martinoli, M.G. Diabetes, a contemporary risk for Parkinson’s disease: Epidemiological and cellular evidences. Front. Aging Neurosci. 2019, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Camargo Maluf, F.; Feder, D.; Alves de Siqueira Carvalho, A. Analysis of the relationship between type II diabetes mellitus and Parkinson’s disease: A systematic review. Parkinsons Dis. 2019, 2019, 4951379. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Sharma Kandel, R.; Mishra, R.; Gautam, J.; Alaref, A.; Jahan, N. Diabetes mellitus and Parkinson’s disease: Shared pathophysiological links and possible therapeutic implications. Cureus 2020, 12, e9853. [Google Scholar] [CrossRef]
- Biosa, A.; Outeiro, T.F.; Bubacco, L.; Bisaglia, M. Diabetes mellitus as a risk factor for Parkinson’s disease: A molecular point of view. Mol. Neurobiol. 2018, 55, 8754–8763. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Willett, W.C.; Ascherio, A. Diet and Parkinson’s disease: A potential role of dairy products in men. Ann. Neurol. 2002, 52, 793–801. [Google Scholar] [CrossRef]
- Park, M.; Ross, G.W.; Petrovitch, H.; White, L.R.; Masaki, K.H.; Nelson, J.S.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Abbott, R.D. Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology 2005, 64, 1047–1051. [Google Scholar] [CrossRef]
- Sääksjärvi, K.; Knekt, P.; Lundqvist, A.; Männistö, S.; Heliövaara, M.; Rissanen, H.; Järvinen, R. A cohort study on diet and the risk of Parkinson’s disease: The role of food groups and diet quality. Br. J. Nutr. 2013, 109, 329–337. [Google Scholar] [CrossRef]
- Kyrozis, A.; Ghika, A.; Stathopoulos, P.; Vassilopoulos, D.; Trichopoulos, D.; Trichopoulou, A. Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur. J. Epidemiol. 2013, 28, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ju, C.; Jiang, H.; Zhang, D. Dairy foods intake and risk of Parkinson’s disease: A dose-response meta-analysis of prospective cohort studies. Eur. J. Epidemiol. 2014, 29, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.C.; Gao, X.; Kim, I.Y.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A. Intake of dairy foods and risk of Parkinson disease. Neurology 2017, 89, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Byberg, L.; Höijer, J.; Kilander, L.; Larsson, S.C. Milk and fermented milk intake and Parkinson’s disease: Cohort study. Nutrients 2020, 12, 2763. [Google Scholar] [CrossRef] [PubMed]
- Matthysse, J.G.; Lisk, D. Residues of diazinon, coumaphos, ciodrin, methoxychlor, and rotenone in cow’s milk from treatments similar to those used for ectoparasite and fly control on dairy cattle, with notes on safety of diazinon and ciodrin to calves. J. Econ. Entomol. 1968, 61, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Chade, A.R.; Kasten, M.; Tanner, C.M. Nongenetic causes of Parkinson’s disease. J. Neural. Transm. Suppl. 2006, 70, 147–151. [Google Scholar]
- Kistner, A.; Krack, P. Parkinson’s disease: No milk today? Front. Neurol. 2014, 5, 172. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; Petrovitch, H.; Masaki, K.H.; Launer, L.J.; Nelson, J.S.; White, L.R.; Tanner, C.M. Midlife milk consumption and substantia nigra neuron density at death. Neurology 2016, 86, 512–519. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Juul, A.; Michaelsen, K.F. High intakes of skimmed milk, but not meat, increase serum IGF-I and IGFBP-3 in eight-year-old boys. Eur. J. Clin. Nutr. 2004, 58, 1211–1216. [Google Scholar] [CrossRef]
- Sluijs, I.; Forouhi, N.G.; Beulens, J.W.; van der Schouw, Y.T.; Agnoli, C.; Arriola, L.; Balkau, B.; Barricarte, A.; Boeing, H.; Bueno-de-Mesquita, H.B.; et al. The amount and type of dairy product intake and incident type 2 diabetes: Results from the EPIC-InterAct Study. Am. J. Clin. Nutr. 2012, 96, 382–390. [Google Scholar]
- Hruby, A.; Ma, J.; Rogers, G.; Meigs, J.B.; Jacques, P.F. Associations of dairy intake with incident prediabetes or diabetes in middle-aged adults vary by both dairy type and glycemic status. J. Nutr. 2017, 147, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chavarro, J.E.; Cao, Y.; Qiu, W.; Mucci, L.; Sesso, H.D.; Stampfer, M.J.; Giovannucci, E.; Pollak, M.; Liu, S.; et al. Whole milk intake is associated with prostate cancer-specific mortality among U.S. male physicians. J. Nutr. 2013, 143, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Brouwer-Brolsma, E.M.; Sluik, D.; Singh-Povel, C.M.; Feskens, E.J.M. Dairy product consumption is associated with pre-diabetes and newly diagnosed type 2 diabetes in the Lifelines Cohort Study. Br. J. Nutr. 2018, 119, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Leucine signaling in the pathogenesis of type 2 diabetes and obesity. World J. Diabetes 2012, 3, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. The pathogenic role of persistent milk signaling in mTORC1- and milk-microRNA-driven type 2 diabetes mellitus. Curr. Diabetes Rev. 2015, 11, 46–62. [Google Scholar] [CrossRef]
- Guillén, C.; Benito, M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front. Endocrinol. 2018, 9, 621. [Google Scholar] [CrossRef]
- Jaafar, R.; Tran, S.; Shah, A.N.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk exosomal miRNAs: Potential drivers of AMPK-to-mTORC1 switching in β-cell de-differentiation of type 2 diabetes mellitus. Nutr. Metab. 2019, 16, 85. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. Metformin: An inhibitor of mTORC1 signaling. J. Endocrinol. Diabetes Obes. 2014, 2, 1029. [Google Scholar]
- Riera-Borrull, M.; García-Heredia, A.; Fernández-Arroyo, S.; Hernández-Aguilera, A.; Cabré, N.; Cuyàs, E.; Luciano-Mateo, F.; Camps, J.; Menendez, J.A.; Joven, J. Metformin potentiates the benefits of dietary restraint: A metabolomic study. Int. J. Mol. Sci. 2017, 18, 2263. [Google Scholar] [CrossRef]
- Howell, J.J.; Ricoult, S.J.; Ben-Sahra, I.; Manning, B.D. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar]
- Noda, T. Regulation of autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef]
- Melnik, B.C.; John, S.M.; Schmitz, G. Milk is not just food but most likely a genetic transfection system activating mTORC1 signaling for postnatal growth. Nutr. J. 2013, 12, 103. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk—A nutrient system of mammalian evolution promoting mTORC1-dependent translation. Int. J. Mol. Sci. 2015, 16, 17048–17087. [Google Scholar] [CrossRef]
- Hoeflich, A.; Meyer, Z. Functional analysis of the IGF-system in milk. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 409–418. [Google Scholar] [CrossRef]
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, X.X.; He, J.; He, S.; Yin, Y. Recent advances in understanding of amino acid signaling to mTORC1 activation. Front. Biosci. Landmark Ed. 2019, 24, 971–982. [Google Scholar] [PubMed]
- Liu, H.; Huang, B.; Xue, S.; Pong, U.K.; Tsang, L.L.; Zhang, X.; Li, G.; Jiang, X. Functional crosstalk between mTORC1/p70S6K pathway and heterochromatin organization in stress-induced senescence of MSCs. Stem Cell Res. Ther. 2020, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.; Broughton, K.S. Insulinotropic effects of whey: Mechanisms of action, recent clinical trials, and clinical applications. Ann. Nutr. Metab. 2016, 69, 56–63. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Dalum, C.; Vaag, A.; Michaelsen, K.F. Differential effects of casein versus whey on fasting plasma levels of insulin, IGF-1 and IGF-1/IGFBP-3: Results from a randomized 7-day supplementation study in prepubertal boys. Eur. J. Clin. Nutr. 2009, 63, 1076–1083. [Google Scholar] [CrossRef]
- Qin, L.Q.; He, K.; Xu, J.Y. Milk consumption and circulating insulin-like growth factor-I level: A systematic literature review. Int. J. Food Sci. Nutr. 2009, 60 (Suppl. 7), 330–340. [Google Scholar] [CrossRef]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys. Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef]
- Lan, A.P.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR signaling in Parkinson’s disease. Neuromolecular Med. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.; Song, J.; Lu, J.; et al. Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. From rapalogs to anti-aging formula. Oncotarget 2017, 8, 35492–35507. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK signaling as a neuroprotective strategy in Parkinson’s disease. J. Parkinsons Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Shaikh, M.F.; Othman, I. Emerging neuroprotective effect of metformin in Parkinson’s disease: A molecular crosstalk. Pharmacol. Res. 2020, 152, 104593. [Google Scholar] [CrossRef]
- Mor, D.E.; Sohrabi, S.; Kaletsky, R.; Keyes, W.; Tartici, A.; Kalia, V.; Miller, G.W.; Murphy, C.T. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 26438–26447. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Perera, G.; Bhadbhade, M.; Halliday, G.M.; Dzamko, N. Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 2019, 294, 14241–14256. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Schwarzschild, M.A. Urate as a marker of risk and progression of neurodegenerative disease. Neurotherapeutics 2017, 14, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Boulos, C.; Yaghi, N.; El Hayeck, R.; Heraoui, G.N.; Fakhoury-Sayegh, N. Nutritional risk factors, microbiota and Parkinson’s disease: What is the current evidence? Nutrients 2019, 11, 1896. [Google Scholar] [CrossRef]
- Sheng, Y.L.; Chen, X.; Hou, X.O.; Yuan, X.; Yuan, B.S.; Yuan, Y.Q.; Zhang, Q.L.; Cao, X.; Liu, C.F.; Luo, W.F.; et al. Urate promotes SNCA/α-synuclein clearance via regulating mTOR-dependent macroautophagy. Exp. Neurol. 2017, 297, 138–147. [Google Scholar] [CrossRef]
- Li, Y.F.; Ouyang, S.H.; Tu, L.F.; Wang, X.; Yuan, W.L.; Wang, G.E.; Wu, Y.P.; Duan, W.J.; Yu, H.M.; Fang, Z.Z.; et al. Caffeine protects skin from oxidative stress-induced senescence through the activation of autophagy. Theranostics 2018, 8, 5713–5730. [Google Scholar] [CrossRef]
- Saiki, S.; Sasazawa, Y.; Imamichi, Y.; Kawajiri, S.; Fujimaki, T.; Tanida, I.; Kobayashi, H.; Sato, F.; Sato, S.; Ishikawa, K.; et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176–187. [Google Scholar] [CrossRef]
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A review of the role of green tea (Camellia sinensis) in antiphotoaging, stress resistance, neuroprotection, and autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Polzella, M.; Frati, A.; Fornai, F. Phytochemicals bridging autophagy induction and alpha-synuclein degradation in parkinsonism. Int. J. Mol. Sci. 2019, 20, 3274. [Google Scholar] [CrossRef]
- Xiao, X.; Shang, X.; Zhai, B.; Zhang, H.; Zhang, T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem. Int. 2018, 114, 58–70. [Google Scholar] [CrossRef]
- Ni, H.; Xu, S.; Chen, H.; Dai, Q. Nicotine modulates CTSS (cathepsin S) synthesis and secretion through regulating the autophagy-lysosomal machinery in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2054–2069. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Lai, M.; Zhang, Y.; Zheng, L.; Xing, Z.; Li, T.; Zou, Z.; Song, Q.; Zhao, X.; Xia, L.; et al. Exosome release is regulated by mTORC1. Adv. Sci. 2018, 6, 1801313. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [PubMed]
- Hassanpour, M.; Rezabakhsh, A.; Rezaie, J.; Nouri, M.; Rahbarghazi, R. Exosomal cargos modulate autophagy in recipient cells via different signaling pathways. Cell Biosci. 2020, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. MicroRNAs: Milk’s epigenetic regulators. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. Milk’s role as an epigenetic regulator in health and disease. Diseases 2017, 5, 12. [Google Scholar] [CrossRef]
- Melnik, B.C. Milk: An epigenetic amplifier of FTO-mediated transcription? Implications for Western diseases. J. Transl. Med. 2015, 13, 385. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. DNA methyltransferase 1-targeting miRNA-148a of dairy milk: A potential bioactive modifier of the human epigenome. Funct. Foods Health Disease 2017, 7, 671–687. [Google Scholar] [CrossRef][Green Version]
- Melnik, B.C.; Kakulas, F. Milk exosomes and microRNAs: Potential epigenetic regulators. In Handbook of Nutrition, Diet, and Epigenetics; Patel, V., Preedy, V., Eds.; Springer: Cham, Switzerland, 2017; pp. 1–28. [Google Scholar]
- Ozkan, H.; Tuzun, F.; Taheri, S.; Korhan, P.; Akokay, P.; Yılmaz, O.; Duman, N.; Özer, E.; Tufan, E.; Kumral, A.; et al. Epigenetic programming through breast milk and its impact on milk-siblings mating. Front. Genet. 2020, 11, 569232. [Google Scholar] [CrossRef]
- Reinhardt, T.A.; Lippolis, J.D.; Nonnecke, B.J.; Sacco, R.E. Bovine milk exosome proteome. J. Proteomics 2012, 75, 1486–1492. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Elbaum Shiff, Y.; Moshayoff, V.; Schecter, D.; Leshkowitz, D.; Reif, S. Characterization and biological function of milk-derived miRNAs. Mol. Nutr. Food Res. 2017, 61, 1700009. [Google Scholar] [CrossRef] [PubMed]
- Zempleni, J.; Aguilar-Lozano, A.; Sadri, M.; Sukreet, S.; Manca, S.; Wu, D.; Zhou, F.; Mutai, E. Biological activities of extracellular vesicles and their cargos from bovine and human milk in humans and implications for infants. J. Nutr. 2017, 147, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Ly, S.; Shan, S.T.; Laugier, J.; Boilard, E.; Gilbert, C.; Provost, P. A subset of extracellular vesicles carries the bulk of microRNAs in commercial dairy cow’s milk. J. Extracell. Vesicles 2017, 6, 1401897. [Google Scholar] [CrossRef]
- Benmoussa, A.; Laugier, J.; Beauparlant, C.J.; Lambert, M.; Droit, A.; Provost, P. Complexity of the microRNA transcriptome of cow milk and milk-derived extracellular vesicles isolated via differential ultracentrifugation. J. Dairy Sci. 2020, 103, 16–29. [Google Scholar] [CrossRef]
- Chen, X.; Gao, C.; Li, H. Identification and characterization of microRNAs in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Res. 2010, 20, 1128–1137. [Google Scholar] [CrossRef]
- Benmoussa, A.; Lee, C.H.; Laffont, B.; Savard, P.; Laugier, J.; Boilard, E.; Gilbert, C.; Fliss, I.; Provost, P. Commercial dairy cow milk microRNAs resist digestion under simulated gastrointestinal tract conditions. J. Nutr. 2016, 146, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Baier, S.R.; Zempleni, J. The intestinal transport of bovine milk exosomes is mediated by endocytosis in human colon carcinoma Caco-2 cells and rat small intestinal IEC-6 cells. J. Nutr. 2015, 145, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Du, X.; Li, J.; Lönnerdal, B. Human milk exosomes and their microRNAs survive digestion in vitro and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2017, 61, 11. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.; Liao, Y.; Du, X.; Xu, W.; Li, J.; Lönnerdal, B. Exosomal microRNAs in milk from mothers delivering preterm infants survive in vitro digestion and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2018, 62, e1701050. [Google Scholar] [CrossRef] [PubMed]
- Kusuma, R.J.; Manca, S.; Friemel, T.; Sukreet, S.; Nguyen, C.; Zempleni, J. Human vascular endothelial cells transport foreign exosomes from cow’s milk by endocytosis. Am. J. Physiol. Cell Physiol. 2016, 310, C800–C807. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hock, A.; Wu, R.Y.; Minich, A.; Botts, S.R.; Lee, C.; Antounians, L.; Miyake, H.; Koike, Y.; Chen, Y.; et al. Bovine milk-derived exosomes enhance goblet cell activity and prevent the development of experimental necrotizing enterocolitis. PLoS ONE 2019, 14, e0211431. [Google Scholar] [CrossRef] [PubMed]
- Reif, S.; Elbaum Shiff, Y.; Golan-Gerstl, R. Milk-derived exosomes (MDEs) have a different biological effect on normal fetal colon epithelial cells compared to colon tumor cells in a miRNA-dependent manner. J. Transl. Med. 2019, 17, 325. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhu, S.; Yuan, M.; Cui, H.; Wang, L.; Luo, X.; Li, J.; Zhou, H.; Tang, Y.; Shen, N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 2010, 184, 6773–6781. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, A.; Rauch, T.A.; Todorov, I.; Ku, H.T.; Al-Abdullah, I.H.; Kandeel, F.; Mullen, Y.; Pfeifer, G.P.; Ferreri, K. Insulin gene expression is regulated by DNA methylation. PLoS ONE 2009, 4, e6953, Erratum in 2009, 4. [Google Scholar] [CrossRef]
- Ouni, M.; Gunes, Y.; Belot, M.P.; Castell, A.L.; Fradin, D.; Bougnères, P. The IGF1 P2 promoter is an epigenetic QTL for circulating IGF1 and human growth. Clin. Epigenetics 2015, 7, 22. [Google Scholar] [CrossRef]
- Ouni, M.; Belot, M.P.; Castell, A.L.; Fradin, D.; Bougnères, P. The P2 promoter of the IGF1 gene is a major epigenetic locus for GH responsiveness. Pharm. J. 2016, 16, 102–106. [Google Scholar] [CrossRef]
- Liu, Z.W.; Zhang, J.T.; Cai, Q.Y.; Zhang, H.X.; Wang, Y.H.; Yan, H.T.; Wu, H.M.; Yang, X.J. Birth weight is associated with placental fat mass- and obesity-associated gene expression and promoter methylation in a Chinese population. J. Matern. Fetal Neonatal. Med. 2016, 29, 106–111. [Google Scholar] [CrossRef]
- Cao, H.; Wang, L.; Chen, B.; Zheng, P.; He, Y.; Ding, Y.; Deng, Y.; Lu, X.; Guo, X.; Zhang, Y.; et al. DNA demethylation upregulated Nrf2 expression in Alzheimer’s disease cellular model. Front. Aging Neurosci. 2016, 7, 244. [Google Scholar] [CrossRef]
- Bendavit, G.; Aboulkassim, T.; Hilmi, K.; Shah, S.; Batist, G. Nrf2 transcription factor can directly regulate mTOR: Linking cytoprotective gene expression to a major metabolic regulator that generates redox activity. J. Biol. Chem. 2016, 291, 25476–25488. [Google Scholar] [CrossRef]
- Manca, S.; Upadhyaya, B.; Mutai, E.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R.; Zempleni, J. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 2018, 8, 11321. [Google Scholar] [CrossRef]
- Jakowec, M.W.; Donaldson, D.M.; Barba, J.; Petzinger, G.M. Postnatal expression of alpha-synuclein protein in the rodent substantia nigra and striatum. Dev. Neurosci. 2001, 23, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The synaptic function of α-synuclein. J. Parkinsons Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef]
- Westphal, C.H.; Chandra, S.S. Monomeric synucleins generate membrane curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Lou, X.; Kim, J.; Hawk, B.J.; Shin, Y.K. α-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J. 2017, 474, 2039–2049. [Google Scholar] [CrossRef]
- Xiong, Q.Y.; Yu, C.; Zhang, Y.; Ling, L.; Wang, L.; Gao, J.L. Key proteins involved in insulin vesicle exocytosis and secretion. Biomed. Rep. 2017, 6, 134–139. [Google Scholar] [CrossRef]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Li, J.D. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Mutai, E.; Zhou, F.; Zempleni, J. Depletion of dietary bovine milk exosomes impairs sensorimotor gating and spatial learning in C57BL/6 mice. FASEB J. 2018, 31, S1. [Google Scholar]
- Ortega-Anaya, J.; Jiménez-Flores, R. Symposium review: The relevance of bovine milk phospholipids in human nutrition—Evidence of the effect on infant gut and brain development. J. Dairy Sci. 2019, 102, 2738–2748. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Properzi, F.; Ferroni, E.; Poleggi, A.; Vinci, R. The regulation of exosome function in the CNS: Implications for neurodegeneration. Swiss Med. Wkly. 2015, 145, w14204. [Google Scholar] [CrossRef]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on extracellular vesicles: Exosomes and their role in protein trafficking and biomarker potential in Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Chistiakov, A.A. α-Synuclein-carrying extracellular vesicles in Parkinson’s disease: Deadly transmitters. Acta Neurol. Belg. 2017, 117, 43–51. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, G.; Lööv, C.; Persson, E.; Lázaro, D.F.; Takeda, S.; Bergström, J.; Erlandsson, A.; Sehlin, D.; Balaj, L.; György, B.; et al. Secretion and uptake of α-synuclein via extracellular vesicles in cultured cells. Cell Mol. Neurobiol. 2018, 38, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Ikenaka, K.; Suzuki, M.; Mochizuki, H.; Nagai, Y. Lipids as trans-acting effectors for α-synuclein in the pathogenesis of Parkinson’s disease. Front. Neurosci. 2019, 13, 693. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Li, R.; Yang, Z.F.; Wang, Y.Z.; Gong, X.L.; Wang, X.M. A DNA methyltransferase inhibitor, 5-aza-2’-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci. Ther. 2013, 19, 183–190. [Google Scholar] [CrossRef]
- Jiang, W.; Li, J.; Zhang, Z.; Wang, H.; Wang, Z. Epigenetic upregulation of alpha-synuclein in the rats exposed to methamphetamine. Eur. J. Pharmacol. 2014, 745, 243–248. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Wu, L.; Zhao, Z.B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.F.; Chen, S.D. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism Relat. Disord. 2014, 20, 308–313. [Google Scholar] [CrossRef]
- Müller, T.; Kohlhepp, W. Hypomethylation in Parkinson’s disease: An epigenetic drug effect? Mov. Disord. 2016, 31, 605. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Kaut, O.; deBoni, L.; Piston, D.; Schmitt, I. DNA methylation in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 108–120. [Google Scholar]
- Guhathakurta, S.; Bok, E.; Evangelista, B.A.; Kim, Y.S. Deregulation of α-synuclein in Parkinson’s disease: Insight from epigenetic structure and transcriptional regulation of SNCA. Prog. Neurobiol. 2017, 154, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Eryilmaz, I.E.; Cecener, G.; Erer, S.; Egeli, U.; Tunca, B.; Zarifoglu, M.; Elibol, B.; Bora Tokcaer, A.; Saka, E.; Demirkiran, M.; et al. Epigenetic approach to early-onset Parkinson’s disease: Low methylation status of SNCA and PARK2 promoter regions. Neurol. Res. 2017, 39, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Furtado, J.C.; Mazurek, M.F. MPTP-induced neurotoxicity and the quest for a preventative therapy for Parkinson’s disease. Can. J. Neurol. Sci. 1991, 18, 77–82. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13 (Suppl. 1), 35–38. [Google Scholar]
- Przedborski, S.; Vila, M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: A tool to explore the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 189–198. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Dovero, S.; Gross, C.; Bezard, E. Unexpected toxicity of very low dose MPTP in mice: A clue to the etiology of Parkinson’s disease? Synapse 2016, 70, 49–51. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, H.; Cheng, J.; Zhu, Y.; Xiao, Q.; Bai, Y.; Tao, J. Neuroprotective effects of lithium on a chronic MPTP mouse model of Parkinson’s disease via regulation of α synuclein methylation. Mol. Med. Rep. 2019, 19, 4989–4997. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Patel, B.M. Crossing the blood-brain barrier: Recent advances in drug delivery to the brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Gosselet, F.; Duban-Deweer, S.; Pottiez, G.; Karamanos, Y. Targeting and crossing the blood-brain barrier with extracellular vesicles. Cells 2020, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Pereira, F.B.; Raimundo, N.; Oliveira, P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic male Parkinson’s disease patients uncovers metabolic and mitochondrial bioenergetic defects. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165615. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017, 8, e3117. [Google Scholar] [CrossRef]
- Fan, R.Z.; Guo, M.; Luo, S.; Cui, M.; Tieu, K. Exosome release and neuropathology induced by α-synuclein: New insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol. Commun. 2019, 7, 184. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; LoGrasso, P.; Lasmézas, C.I. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Nagoor Meeran, M.F.; Azimullah, S.; Adem, A.; Sadek, B.; Ojha, S.K. Plant extracts and phytochemicals targeting α-synuclein aggregation in Parkinson’s disease models. Front. Pharmacol. 2019, 9, 1555. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Sala, G.; Marinig, D.; Arosio, A.; Ferrarese, C. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s sisease. Front. Mol. Neurosci. 2016, 9, 157. [Google Scholar] [CrossRef]
- Campbell, P.; Morris, H.; Schapira, A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert Opin. Ther. Targets 2018, 22, 823–832. [Google Scholar] [CrossRef]
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Klaver, A.C.; Coffey, M.P.; Aasly, J.O.; Loeffler, D.A. CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res. 2018, 1683, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Mao, Z. Dysregulation of autophagy and Parkinson’s disease: The MEF2D link. Apoptosis 2010, 15, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Yang, X.; Lou, J. Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. 2016, 1644, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Ding, L. Downregulation of miR-21 suppresses 1-methyl-4-phenylpyridinium-induced neuronal damage in MES23.5 cells. Exp. Ther. Med. 2019, 18, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhen, J.; Lu, Z. Synergetic neuroprotective effect of docosahexaenoic acid and aspirin in SH-Y5Y by inhibiting miR-21 and activating RXRα and PPARα. DNA Cell Biol. 2017, 36, 482–489. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Kirik, D.; Stefanis, L. LAMP2A as a therapeutic target in Parkinson disease. Autophagy 2013, 9, 2166–2168. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; John, S.M.; Carrera-Bastos, P.; Schmitz, G. MicroRNA-21-enriched exosomes as epigenetic regulators in melanomagenesis and melanoma progression: The impact of Western lifestyle factors. Cancers 2020, 12, 2111. [Google Scholar] [CrossRef]
- Masoudi, M.S.; Mehrabian, E.; Mirzaei, H. MiR-21: A key player in glioblastoma pathogenesis. J. Cell Biochem. 2018, 119, 1285–1290. [Google Scholar] [CrossRef]
- Ye, Q.; Wen, Y.; Al-Kuwari, N.; Chen, X. Association between Parkinson’s disease and melanoma: Putting the pieces together. Front. Aging Neurosci. 2020, 12, 60. [Google Scholar] [CrossRef]
- Mencke, P.; Hanss, Z.; Boussaad, I.; Sugier, P.E.; Elbaz, A.; Krüger, R. Bidirectional relation between Parkinson’s disease and glioblastoma multiforme. Front. Neurol. 2020, 11, 898. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. Exosomes of pasteurized milk: Potential pathogens of Western diseases. J. Transl. Med. 2019, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sadri, M.; Giraud, D.; Zempleni, J. RNase H2-dependent polymerase chain reaction and elimination of confounders in sample collection, storage, and analysis strengthen evidence that microRNAs in bovine milk are bioavailable in humans. J. Nutr. 2018, 148, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Ge, S.; Sun, J. Ailanthone exerts anticancer effect by up-regulating miR-148a expression in MDA-MB-231 breast cancer cells and inhibiting proliferation, migration and invasion. Biomed. Pharmacother. 2019, 109, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, V.; Danzer, K.M. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 2018, 373, 175–182. [Google Scholar] [CrossRef]
- Yu, S.; Zhao, Z.; Sun, L.; Li, P. Fermentation results in quantitative changes in milk-derived exosomes and different effects on cell growth and survival. J. Agric. Food Chem. 2017, 65, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Grote, V.; Verduci, E.; Scaglioni, S.; Vecchi, F.; Contarini, G.; Giovannini, M.; Koletzko, B.; Agostoni, C.; European Childhood Obesity Project. Breast milk composition and infant nutrient intakes during the first 12 months of life. Eur. J. Clin. Nutr. 2016, 70, 250–256. [Google Scholar] [CrossRef]
- Kunst, C.; Kliegman, R.; Trindade, C. The glucose-galactose paradox in neonatal murine hepatic glycogen synthesis. Am. J. Physiol. 1989, 257, E697–E703. [Google Scholar] [CrossRef]
- Brown, L.D.; Cavalli, C.; Harwood, J.E.; Casadei, A.; Teng, C.C.; Traggiai, C.; Serra, G.; Bevilacqua, G.; Battaglia, F.C. Plasma concentrations of carbohydrates and sugar alcohols in term newborns after milk feeding. Pediatr. Res. 2008, 64, 189–193. [Google Scholar] [CrossRef]
- Spedale, S.B.; Battaglia, F.C.; Sparks, J.W. Hepatic metabolism of glucose, galactose, and lactate after milk feeding in newborn lambs. Am. J. Physiol. 1992, 262 Pt 1, E46–E51. [Google Scholar] [CrossRef]
- Henderson, J.M.; Kutner, M.H.; Bain, R.P. First-order clearance of plasma galactose: The effect of liver disease. Gastroenterology 1982, 83, 1090–1096. [Google Scholar] [CrossRef]
- Lange, A.; Grønbæk, H.; Vilstrup, H.; Keiding, S. Age-dependency of galactose elimination capacity in healthy children and children with chronic liver disease. Scand. J. Gastroenterol. 2011, 46, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bua, V.; Brunori, A.; Bianchi, G.; Pisi, P.; Fabbri, A.; Zoli, M.; Pisi, E. Galactose elimination capacity and liver volume in aging man. Hepatology 1988, 8, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Schnegg, M.; Lauterburg, B.H. Quantitative liver function in the elderly assessed by galactose elimination capacity, aminopyrine demethylation and caffeine clearance. J. Hepatol. 1986, 3, 164–171. [Google Scholar] [CrossRef]
- Jepsen, P.; Vilstrup, H.; Ott, P.; Keiding, S.; Andersen, P.K.; Tygstrup, N. The galactose elimination capacity and mortality in 781 Danish patients with newly-diagnosed liver cirrhosis: A cohort study. BMC Gastroenterol. 2009, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Michaëlsson, K.; Wolk, A.; Langenskiöld, S.; Basu, S.; Warensjö Lemming, E.; Melhus, H.; Byberg, L. Milk intake and risk of mortality and fractures in women and men: Cohort studies. BMJ 2014, 349, g6015. [Google Scholar] [CrossRef]
- Michaëlsson, K.; Byberg, L. Mixing of apples and oranges in milk research: A cohort analysis of non-fermented milk intake and all-cause mortality. Nutrients 2020, 12, 1393. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; Nakamura, K. Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett. 2015, 589, 3702–3713. [Google Scholar] [CrossRef] [PubMed]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Hunn, B.H.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Nicklas, W.J.; Youngster, S.K.; Kindt, M.V.; Heikkila, R.E. MPTP, MPP+ and mitochondrial function. Life Sci. 1987, 40, 721–729. [Google Scholar] [CrossRef]
- Przedborski, S.; Tieu, K.; Perier, C.; Vila, M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J. Bioenerg. Biomembr. 2004, 36, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Telford, J.E.; Kilbride, S.M.; Davey, G.P. Complex I is rate-limiting for oxygen consumption in the nerve terminal. J. Biol. Chem. 2009, 284, 9109–9114. [Google Scholar] [CrossRef]
- Cui, X.; Zuo, P.; Zhang, Q.; Li, X.; Hu, Y.; Long, J.; Packer, L.; Liu, J. Chronic systemic D-galactose exposure induces memory loss, neurodegeneration, and oxidative damage in mice: Protective effects of R-alpha-lipoic acid. J. Neurosci. Res. 2006, 84, 647–654. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Majdi, A.; McCann, S.K.; Mahmoudi, J.; Vafaee, M.S.; Macleod, M.R. D-galactose-induced brain ageing model: A systematic review and meta-analysis on cognitive outcomes and oxidative stress indices. PLoS ONE 2017, 12, e0184122. [Google Scholar]
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of D-galactose-induced brain aging and its potential used for therapeutic interventions. Exp. Gerontol. 2018, 101, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yao, H.; Chen, X.; Wang, Z.; Xiang, Y.; Xia, J.; Liu, Y.; Wang, Y. Ginsenoside Rg1 decreases oxidative stress and down-regulates Akt/mTOR signaling to attenuate cognitive impairment in mice and senescence of neural stem cells induced by D-galactose. Neurochem. Res. 2018, 43, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhong, Y.; Peng, W.; Sun, Y.; Hu, Y.J.; Yang, Y.; Kong, W.J. Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of D-galactose-induced aging rats. Mol. Biol. Rep. 2011, 38, 3635–3642. [Google Scholar] [CrossRef] [PubMed]
- González-Casacuberta, I.; Juárez-Flores, D.L.; Ezquerra, M.; Fucho, R.; Catalán-García, M.; Guitart-Mampel, M.; Tobías, E.; García-Ruiz, C.; Fernández-Checa, J.C.; Tolosa, E.; et al. Mitochondrial and autophagic alterations in skin fibroblasts from Parkinson disease patients with Parkin mutations. Aging 2019, 11, 3750–3767. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Flores, D.L.; González-Casacuberta, I.; Ezquerra, M.; Bañó, M.; Carmona-Pontaque, F.; Catalán-García, M.; Guitart-Mampel, M.; Rivero, J.J.; Tobias, E.; Milisenda, J.C.; et al. Exhaustion of mitochondrial and autophagic reserve may contribute to the development of LRRK2 G2019S -Parkinson’s disease. J. Transl. Med. 2018, 16, 160. [Google Scholar] [CrossRef]
- Bei, Y.; Wu, X.; Cretoiu, D.; Shi, J.; Zhou, Q.; Lin, S.; Wang, H.; Cheng, Y.; Zhang, H.; Xiao, J.; et al. miR-21 suppression prevents cardiac alterations induced by d-galactose and doxorubicin. J. Mol. Cell Cardiol. 2018, 115, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Pan, B.; Zhou, Z.; Zhou, L.; Li, Z.; Zhang, Z. MicroRNA-21 regulates cell proliferation and apoptosis in H₂O₂-stimulated rat spinal cord neurons. Mol. Med. Rep. 2015, 12, 7011–7016. [Google Scholar] [CrossRef][Green Version]
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc. Diabetol. 2018, 17, 105. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef]
- Perfeito, R.; Lázaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Brahmachari, S.; Karuppagounder, S.S.; Ge, P.; Lee, S.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. c-Abl and Parkinson’s disease: Mechanisms and therapeutic potential. J. Parkinsons Dis. 2017, 7, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Gonfloni, S.; Maiani, E.; Di Bartolomeo, C.; Diederich, M.; Cesareni, G. Oxidative stress, DNA damage, and c-Abl signaling: At the crossroad in neurodegenerative diseases? Int. J. Cell Biol. 2012, 2012, 683097. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Ohtsuki, S.; Couraud, P.O.; Suzuki, T.; Terasaki, T. Oxidative stress-induced activation of Abl and Src kinases rapidly induces P-glycoprotein internalization via phosphorylation of caveolin-1 on tyrosine-14, decreasing cortisol efflux at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2020, 40, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.R.; Sandoval, P.C.; Leal, N.R.; Castro, P.U.; Kosik, K.S. Activation of the neuronal c-Abl tyrosine kinase by amyloid-beta-peptide and reactive oxygen species. Neurobiol. Dis. 2004, 17, 326–336. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. c-Abl phosphorylates α-synuclein and regulates its degradation: Implication for α-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Aspholm, E.E.; Matečko-Burmann, I.; Burmann, B.M. Keeping α-synuclein at bay: A more active role of molecular chaperones in preventing mitochondrial interactions and transition to pathological states? Life 2020, 10, 289. [Google Scholar] [CrossRef]
- Ohlsson, J.A.; Johansson, M.; Hansson, H.; Abrahamson, A.; Byberg, L.; Smedman, A.; Lindmark-Månsson, H.; Lund, Å. Lactose, glucose and galactose content in milk, fermented milk and lactose-free milk products. Int. Dairy J. 2017, 73, 151–154. [Google Scholar] [CrossRef]
- Portnoi, P.A.; MacDonald, A. The lactose and galactose content of cheese suitable for galactosaemia: New analysis. JIMD Rep. 2016, 29, 85–87. [Google Scholar] [PubMed]
- Van Calcar, S.C.; Bernstein, L.E.; Rohr, F.J.; Yannicelli, S.; Berry, G.T.; Scaman, C.H. Galactose content of legumes, caseinates, and some hard cheeses: Implications for diet treatment of classic galactosemia. J. Agric. Food Chem. 2014, 62, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- O’leary, V.S.; Woychik, J.H. Utilization of lactose, glucose, and galactose by a mixed culture of Streptococcus thermophilus and Lactobacillus bulgaricus in milk treated with lactase enzyme. Appl. Environ. Microbiol. 1976, 32, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kandler, O. Carbohydrate metabolism in lactic acid bacteria. Antonie Leeuwenhoek 1983, 49, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Konings, W.N.; Poolman, B.; Driessen, A.J. Bioenergetics and solute transport in lactococci. Crit. Rev. Microbiol. 1989, 16, 419–476. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.D.; Turner, K.W.; Crow, V.L. Galactose fermentation by Streptococcus lactis and Streptococcus cremoris: Pathways, products, and regulation. J. Bacteriol. 1980, 144, 672–682. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Bennett, R.D. Role of diet and nutritional supplements in Parkinson’s disease progression. Oxid. Med. Cell Longev. 2017, 2017, 6405278. [Google Scholar] [CrossRef]
- Lenders, C.M.; Liu, S.; Wilmore, D.W.; Sampson, L.; Dougherty, L.W.; Spiegelman, D.; Willett, W.C. Evaluation of a novel food composition database that includes glutamine and other amino acids derived from gene sequencing data. Eur. J. Clin. Nutr. 2009, 63, 1433–1439. [Google Scholar] [CrossRef]
- Mor, D.E.; Murphy, C.T. Mitochondrial hyperactivity as a potential therapeutic target in Parkinson’s disease. Transl. Med. Aging 2020, 4, 117–120. [Google Scholar] [CrossRef]
- Yao, V.; Kaletsky, R.; Keyes, W.; Mor, D.E.; Wong, A.K.; Sohrabi, S.; Murphy, C.T.; Troyanskaya, O.G. An integrative tissue-network approach to identify and test human disease genes. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Mansfeld, J.; Urban, N.; Priebe, S.; Groth, M.; Frahm, C.; Hartmann, N.; Gebauer, J.; Ravichandran, M.; Dommaschk, A.; Schmeisser, S.; et al. Branched-chain amino acid catabolism is a conserved regulator of physiological ageing. Nat. Commun. 2015, 6, 10043. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A future therapy for neurodegenerative diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; He, T.; Zhang, W.; Zhang, G.; Ma, X. Branched chain amino acids: Beyond nutrition metabolism. Int. J. Mol. Sci. 2018, 19, 954. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Qi, Q.; Hruby, A.; Manson, J.E.; Willett, W.C.; Wolpin, B.M.; Hu, F.B.; Qi, L. Cumulative consumption of branched-chain amino acids and incidence of type 2 diabetes. Int. J. Epidemiol. 2016, 45, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Scott, R.A.; Sharp, S.J.; Burgess, S.; Luan, J.; Tillin, T.; Schmidt, A.F.; Imamura, F.; Stewart, I.D.; Perry, J.R.; et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: A mendelian randomisation analysis. PLoS Med. 2016, 13, e1002179. [Google Scholar] [CrossRef]
- Melnik, B.C.; Schmitz, G. Milk consumption does not prevent but induces type 2 diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3200. [Google Scholar] [CrossRef]
- Fontana, L.; Cummings, N.E.; Arriola Apelo, S.I.; Neuman, J.C.; Kasza, I.; Schmidt, B.A.; Cava, E.; Spelta, F.; Tosti, V.; Syed, F.A.; et al. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Rep. 2016, 16, 520–530. [Google Scholar] [CrossRef]
- Cummings, N.E.; Williams, E.M.; Kasza, I.; Konon, E.N.; Schaid, M.D.; Schmidt, B.A.; Poudel, C.; Sherman, D.S.; Yu, D.; Arriola Apelo, S.I.; et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J. Physiol. 2018, 596, 623–645. [Google Scholar] [CrossRef]
- Anderson, J.G.; Hintze, K.; Marchant, E.D. Restricting branched-chain amino acids: An approach to improve metabolic health. J. Physiol. 2018, 596, 2469–2470. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Cogger, V.C.; Pulpitel, T.; Wahl, D.; Clark, X.; Bagley, E.; Gregoriou, G.C.; Senior, A.M.; Wang, Q.P.; Brandon, A.E.; et al. Branched chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat. Metab. 2019, 1, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Munch, E.M.; Harris, R.A.; Mohammad, M.; Benham, A.L.; Pejerrey, S.M.; Showalter, L.; Hu, M.; Shope, C.D.; Maningat, P.D.; Gunaratne, P.H.; et al. Transcriptome profiling of microRNA by Next-Gen deep sequencing reveals known and novel miRNA species in the lipid fraction of human breast milk. PLoS ONE 2013, 8, e50564. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, A.; Provost, P. Milk microRNAs in health and disease. Compr. Rev. Food Sci. Food Saf. 2019, 18, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Dudemaine, P.L.; Li, R.; Ibeagha-Awemu, E.M. Co-expression network and pathway analyses reveal important modules of miRNAs regulating milk yield and component traits. Int. J. Mol. Sci. 2017, 18, 1560. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luo, J.; Sun, S.; Cao, D.; Shi, H.; Loor, J.J. miR-148a and miR-17-5p synergistically regulate milk TAG synthesis via PPARGC1A and PPARA in goat mammary epithelial cells. RNA Biol. 2017, 14, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Konnova, E.A.; Swanberg, M. Animal models of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 5. [Google Scholar]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Hasegawa, K.; Yasuda, T.; Shiraishi, C.; Fujiwara, K.; Przedborski, S.; Mochizuki, H.; Yoshikawa, K. Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat. Commun. 2016, 7, 10943. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.; Huang, W.; Huang, M.; Wang, J.; Chen, X.; Ye, Q. Beneficial effects of PGC-1α in the substantia nigra of a mouse model of MPTP-induced dopaminergic neurotoxicity. Aging 2019, 11, 8937–8950. [Google Scholar] [CrossRef]
- Ye, Q.; Huang, W.; Li, D.; Si, E.; Wang, J.; Wang, Y.; Chen, C.; Chen, X. Overexpression of PGC-1α influences mitochondrial signal transduction of dopaminergic neurons. Mol. Neurobiol. 2016, 53, 3756–3770. [Google Scholar] [CrossRef]
- Ye, Q.; Chen, C.; Si, E.; Cai, Y.; Wang, J.; Huang, W.; Li, D.; Wang, Y.; Chen, X. Mitochondrial effects of PGC-1alpha silencing in MPP+ treated human SH-SY5Y neuroblastoma cells. Front. Mol. Neurosci. 2017, 10, 164. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.; Kim, Y.K.; Shin, D.; Kim, K.S. MPC1 is essential for PGC-1α-induced mitochondrial respiration and biogenesis. Biochem. J. 2018, 475, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Imamura, H.; Sasaoka, N.; Yamamoto, M.; Uemura, N.; Shudo, T.; Fuchigami, T.; Takahashi, R.; Kakizuka, A. ATP maintenance via two types of ATP regulators mitigates pathological phenotypes in mouse models of Parkinson’s disease. EBioMedicine 2017, 22, 225–241. [Google Scholar] [CrossRef]
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H.S. SQSTM1/p62 and PPARGC1A/PGC-1alpha at the interface of autophagy and vascular senescence. Autophagy 2020, 16, 1092–1110. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar]
- Geetha, T.; Vishwaprakash, N.; Sycheva, M.; Babu, J.R. Sequestosome 1/p62: Across diseases. Biomarkers 2012, 17, 99–103. [Google Scholar] [CrossRef]
- Ma, S.; Attarwala, I.Y.; Xie, X.Q. SQSTM1/p62: A potential target for neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef]
- Kalogeropulou, A.F.; Zhao, J.; Bolliger, M.F.; Memou, A.; Narasimha, S.; Molitor, T.P.; Wilson, W.H.; Rideout, H.J.; Nichols, R.J. P62/SQSTM1 is a novel leucine-rich repeat kinase 2 (LRRK2) substrate that enhances neuronal toxicity. Biochem. J. 2018, 475, 1271–1293. [Google Scholar] [CrossRef]
- Guay, C.; Menoud, V.; Rome, S.; Regazzi, R. Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun. Signal. 2015, 13, 17. [Google Scholar] [CrossRef]
- Osmai, M.; Osmai, Y.; Bang-Berthelsen, C.H.; Pallesen, E.M.; Vestergaard, A.L.; Novotny, G.W.; Pociot, F.; Mandrup-Poulsen, T. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes Metab. Res. Rev. 2016, 32, 334–349. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 137–146. [Google Scholar] [CrossRef] [PubMed]
- Jalabert, A.; Vial, G.; Guay, C.; Wiklander, O.P.; Nordin, J.Z.; Aswad, H.; Forterre, A.; Meugnier, E.; Pesenti, S.; Regazzi, R.; et al. Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia 2016, 59, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Xie, X.X.; Xiao, X.; Li, X. Exosome-like vesicles as new mediators and therapeutic targets for treating insulin resistance and β-cell mass failure in type 2 diabetes mellitus. J. Diabetes Res. 2019, 2019, 3256060. [Google Scholar] [CrossRef] [PubMed]
- Javeed, N.; Sagar, G.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Bhattacharya, S.; Truty, M.; Petersen, G.M.; Kaufman, R.J.; Chari, S.T.; et al. Pancreatic cancer-derived exosomes cause paraneoplastic β-cell dysfunction. Clin. Cancer Res. 2015, 21, 1722–1733. [Google Scholar] [CrossRef]
- Lausier, J.; Diaz, W.C.; Roskens, V.; LaRock, K.; Herzer, K.; Fong, C.G.; Latour, M.G.; Peshavaria, M.; Jetton, T.L. Vagal control of pancreatic ß-cell proliferation. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E786–E793. [Google Scholar] [CrossRef]
- Medina, A.; Yamada, S.; Hara, A.; Hamamoto, K.; Kojima, I. Involvement of the parasympathetic nervous system in the initiation of regeneration of pancreatic β-cells. Endocr. J. 2013, 60, 687–696. [Google Scholar] [CrossRef]
- Surguchov, A. Association between type-2 diabetes and Parkinson´s disease: A cross-talk between amylin and α-synuclein. Diabetes Metabol. Syndr. Clin. Res. Rev. 2016, 1, 1–7. [Google Scholar]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Amat-Villegas, I.; Valenti-Azcarate, R.; Carmona-Abellan, M.D.M.; Marcilla, I.; Tuñon, M.T.; Luquin, M.R. Interaction of amyloidogenic proteins in pancreatic β cells from subjects with synucleinopathies. Acta Neuropathol. 2018, 135, 877–886. [Google Scholar] [CrossRef]
- Geng, X.; Lou, H.; Wang, J.; Li, L.; Swanson, A.L.; Sun, M.; Beers-Stolz, D.; Watkins, S.; Perez, R.G.; Drain, P. α-Synuclein binds the K(ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E276–E286. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Martinez, G.; Yang, B.; Vargas-Medrano, J.; Perez, R.G. Could α-synuclein modulation of insulin and dopamine identify a novel link between Parkinson’s disease and diabetes as well as potential therapies? Front. Mol. Neurosci. 2018, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Aamodt, K.I.; Powers, A.C. Signals in the pancreatic islet microenvironment influence β-cell proliferation. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 124–136. [Google Scholar] [CrossRef] [PubMed]
- Moullé, V.S.; Tremblay, C.; Castell, A.L.; Vivot, K.; Ethier, M.; Fergusson, G.; Alquier, T.; Ghislain, J.; Poitout, V. The autonomic nervous system regulates pancreatic β-cell proliferation in adult male rats. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E234–E243. [Google Scholar] [CrossRef]
- Yamamoto, J.; Imai, J.; Izumi, T.; Takahashi, H.; Kawana, Y.; Takahashi, K.; Kodama, S.; Kaneko, K.; Gao, J.; Uno, K.; et al. Neuronal signals regulate obesity induced β-cell proliferation by FoxM1 dependent mechanism. Nat. Commun. 2017, 8, 1930. [Google Scholar] [CrossRef]
- Mucibabic, M.; Steneberg, P.; Lidh, E.; Straseviciene, J.; Ziolkowska, A.; Dahl, U.; Lindahl, E.; Edlund, H. α-Synuclein promotes IAPP fibril formation in vitro and β-cell amyloid formation in vivo in mice. Sci. Rep. 2020, 10, 20438. [Google Scholar] [CrossRef]
- Lubaczeuski, C.; Balbo, S.L.; Ribeiro, R.A.; Vettorazzi, J.F.; Santos-Silva, J.C.; Carneiro, E.M.; Bonfleur, M.L. Vagotomy ameliorates islet morphofunction and body metabolic homeostasis in MSG-obese rats. Braz. J. Med. Biol. Res. 2015, 48, 447–457. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martínez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garican, C.; et al. Pancreatic beta cell death: Novel potential mechanisms in diabetes therapy. J. Diabetes Res. 2018, 2018, 9601801. [Google Scholar] [CrossRef] [PubMed]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M.; Stolarczyk, E.; Tobin, V. The role of GLUT2 in dietary sugar handling. J. Physiol. Biochem. 2005, 61, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Zhao, T.Q. Effects of puerarin on D-galactose-induced memory deficits in mice. Acta Pharmacol. Sin. 2002, 23, 587–590. [Google Scholar]
- Li, L.; Ng, T.B.; Gao, W.; Li, W.; Fu, M.; Niu, S.M.; Zhao, L.; Chen, R.R.; Liu, F. Antioxidant activity of gallic acid from rose flowers in senescence accelerated mice. Life Sci. 2005, 77, 230–240. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, Y.L.; Luo, L.; Wu, D.M.; Sun, D.X.; Feng, Y.J. Quercetin reverses D-galactose induced neurotoxicity in mouse brain. Behav. Brain Res. 2006, 171, 251–260. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, Y.L.; Wu, D.M.; Luo, L.; Sun, D.X.; Shan, Q. Ursolic acid ameliorates cognition deficits and attenuates oxidative damage in the brain of senescent mice induced by D-galactose. Biochem. Pharmacol. 2007, 74, 1078–1090. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Fan, S.H.; Zheng, Y.L.; Lu, J.; Wu, D.M.; Shan, Q.; Hu, B. Purple sweet potato color attenuates oxidative stress and inflammatory response induced by d-galactose in mouse liver. Food Chem. Toxicol. 2009, 47, 496–501. [Google Scholar] [CrossRef]
- Yang, Y.C.; Lin, H.Y.; Su, K.Y.; Chen, C.H.; Yu, Y.L.; Lin, C.C.; Yu, S.L.; Yan, H.Y.; Su, K.J.; Chen, Y.L. Rutin, a flavonoid that is a main component of Saussurea involucrata, attenuates the senescence effect in D-galactose aging mouse model. Evid. Based Complement Alternat. Med. 2012, 2012, 980276. [Google Scholar] [CrossRef]
- Ahangarpour, A.; Oroojan, A.A.; Badavi, M. Exendin-4 protects mice from D-galactose-induced hepatic and pancreatic dysfunction. Pathobiol. Aging Age Relat. Dis. 2017, 8, 1418593. [Google Scholar] [CrossRef] [PubMed]
- Saeedi Borujeni, M.J.; Esfandiary, E.; Baradaran, A.; Valiani, A.; Ghanadian, M.; Codoñer-Franch, P.; Basirat, R.; Alonso-Iglesias, E.; Mirzaei, H.; Yazdani, A. Molecular aspects of pancreatic β-cell dysfunction: Oxidative stress, microRNA, and long noncoding RNA. J. Cell. Physiol. 2019, 234, 8411–8425. [Google Scholar] [CrossRef] [PubMed]
- Barlow, A.D.; Thomas, D.C. Autophagy in diabetes: β-cell dysfunction, insulin resistance, and complications. DNA Cell Biol. 2015, 34, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; García-Macia, M.; Singh, R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 2017, 13, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Mukhopadhyay, M.; Bhattacharyya, M.; Karmakar, P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018, 17, 709–720. [Google Scholar]
- Swiss, R.; Will, Y. Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose- or galactose-containing media. Curr. Protoc. Toxicol. 2011, 49, 2–20. [Google Scholar]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab. 2017, 25, 883–897, Erratum in 2017, 25, 1207. [Google Scholar] [CrossRef]
- Veneri, D.; Franchini, M.; Bonora, E. Imatinib and regression of type 2 diabetes. N. Engl. J. Med. 2005, 352, 1049–1050. [Google Scholar] [CrossRef]
- Couzin, J. Diabetes research. Researchers puzzle over possible effect of Gleevec. Science 2005, 307, 1711. [Google Scholar] [CrossRef]
- Xia, C.Q.; Zhang, P.; Li, S.; Yuan, L.; Xia, T.; Xie, C.; Clare-Salzler, M.J. C-Abl inhibitor imatinib enhances insulin production by β cells: C-Abl negatively regulates insulin production via interfering with the expression of NKx2.2 and GLUT-2. PLoS ONE 2014, 9, e97694. [Google Scholar] [CrossRef]
- Kumar, M.; Kulshrestha, R.; Singh, N.; Jaggi, A.S. Expanding spectrum of anticancer drug, imatinib, in the disorders affecting brain and spinal cord. Pharmacol. Res. 2019, 143, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Welsh, N. Does the small tyrosine kinase inhibitor imatinib mesylate counteract diabetes by affecting pancreatic islet amyloidosis and fibrosis? Expert Opin. Investig. Drugs 2012, 21, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Kurochkin, I.V. Insulin-degrading enzyme: Embarking on amyloid destruction. Trends Biochem. Sci. 2001, 26, 421–425. [Google Scholar] [CrossRef]
- Steneberg, P.; Bernardo, L.; Edfalk, S.; Lundberg, L.; Backlund, F.; Ostenson, C.G.; Edlund, H. The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of α-synuclein levels in β-cells. Diabetes 2013, 62, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chorell, E.; Steneberg, P.; Vernersson-Lindahl, E.; Edlund, H.; Wittung-Stafshede, P. Insulin-degrading enzyme prevents α-synuclein fibril formation in a nonproteolytical manner. Sci. Rep. 2015, 5, 12531. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggrgate formation by CSF exosomes from patients with Parkinson´s disease and dementia with Lewy bodies. Brain 2016, 139, 481–494. [Google Scholar] [CrossRef]
- Hill, A.F. Extracellular vesicles and neurodegenerative diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Beeraka, N.M.; Doreswamy, S.H.; Sadhu, S.P.; Srinivasan, A.; Pragada, R.R.; Madhunapantula, S.V.; Aliev, G. The role of exosomes in stemness and neurodegenerative diseases-chemoresistant-cancer therapeutics and phytochemicals. Int. J. Mol. Sci. 2020, 21, 6818. [Google Scholar] [CrossRef]
- Pardo, F.; Villalobos-Labra, R.; Sobrevia, B.; Toledo, F.; Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Aspects Med. 2018, 60, 81–91. [Google Scholar] [CrossRef]
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetologia 2019, 62, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Castaño, C.; Novials, A.; Párrizas, M. Exosomes and diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3107. [Google Scholar] [CrossRef] [PubMed]
- Gasparyan, A.Y.; Ayvazyan, L.; Blackmore, H.; Kitas, G.D. Writing a narrative biomedical review: Considerations for authors, peer reviewers, and editors. Rheumatol. Int. 2011, 31, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Saracci, C.; Mahamat, M.; Jacquérioz, F. Comment rédiger un article scientifique de type revue narrative de la littérature? [How to write a narrative literature review article?]. Rev. Med. Suisse 2019, 15, 1694–1698. [Google Scholar] [PubMed]




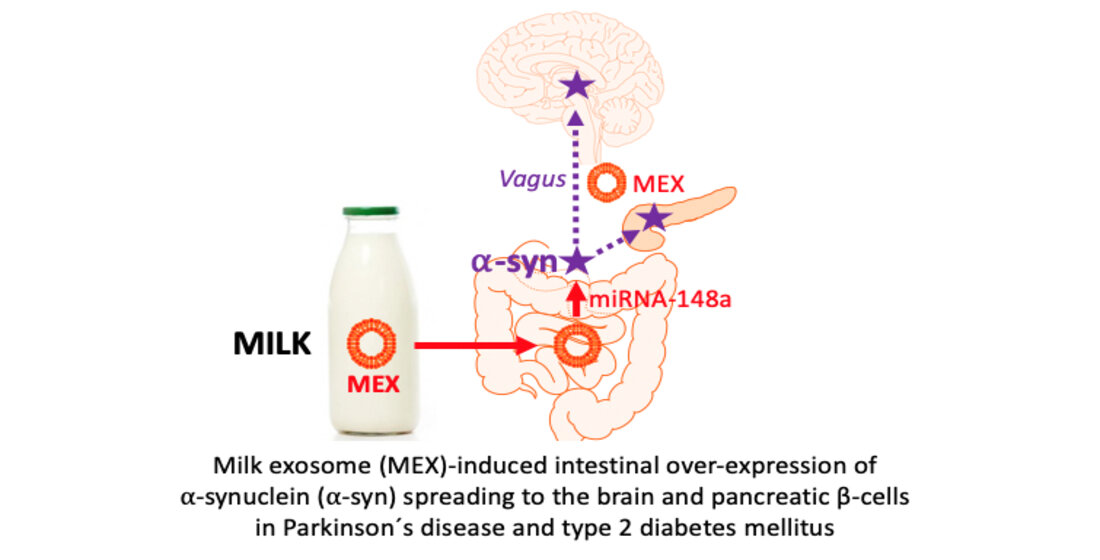{kind=link}
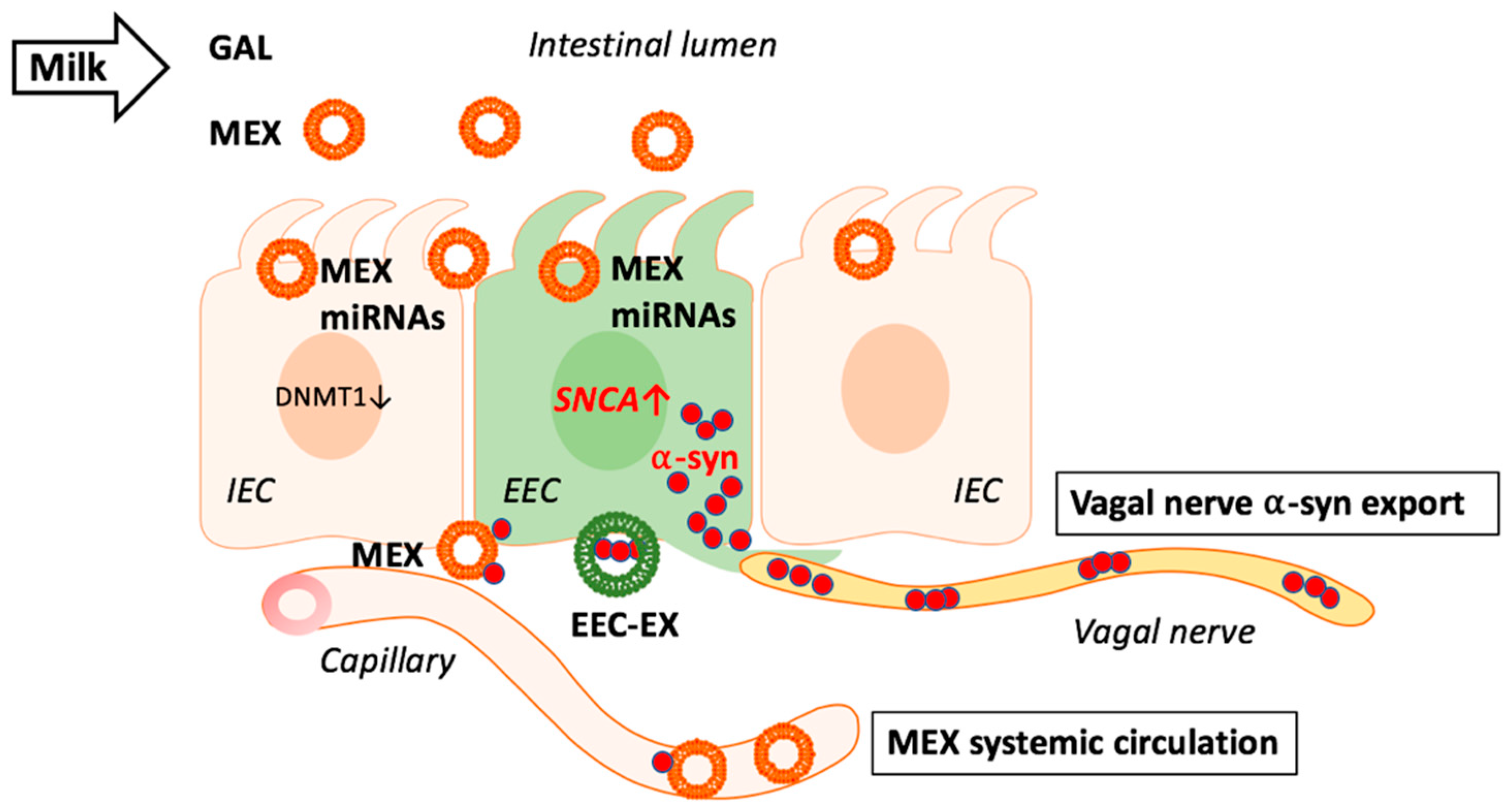{kind=link}
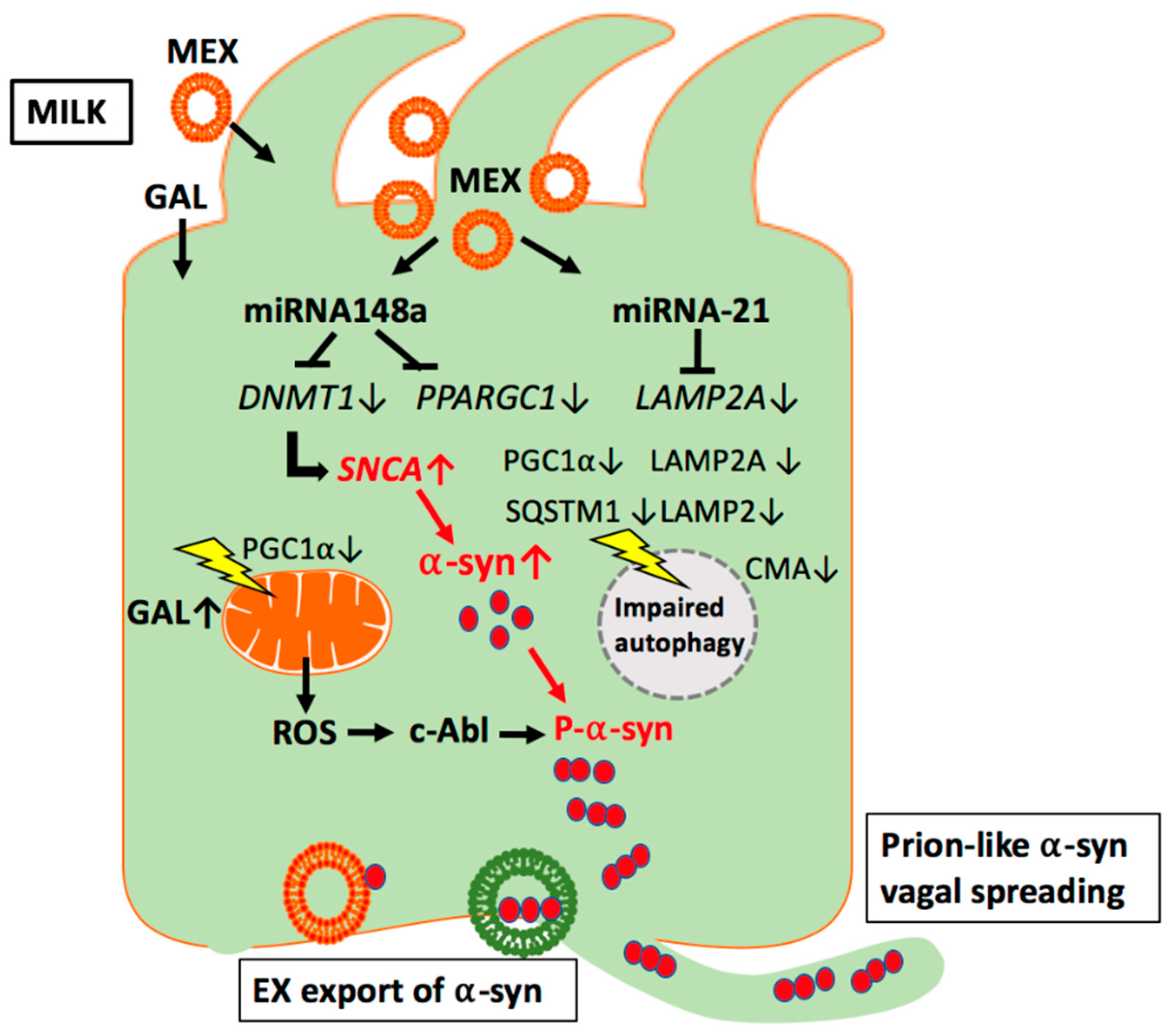{kind=link}
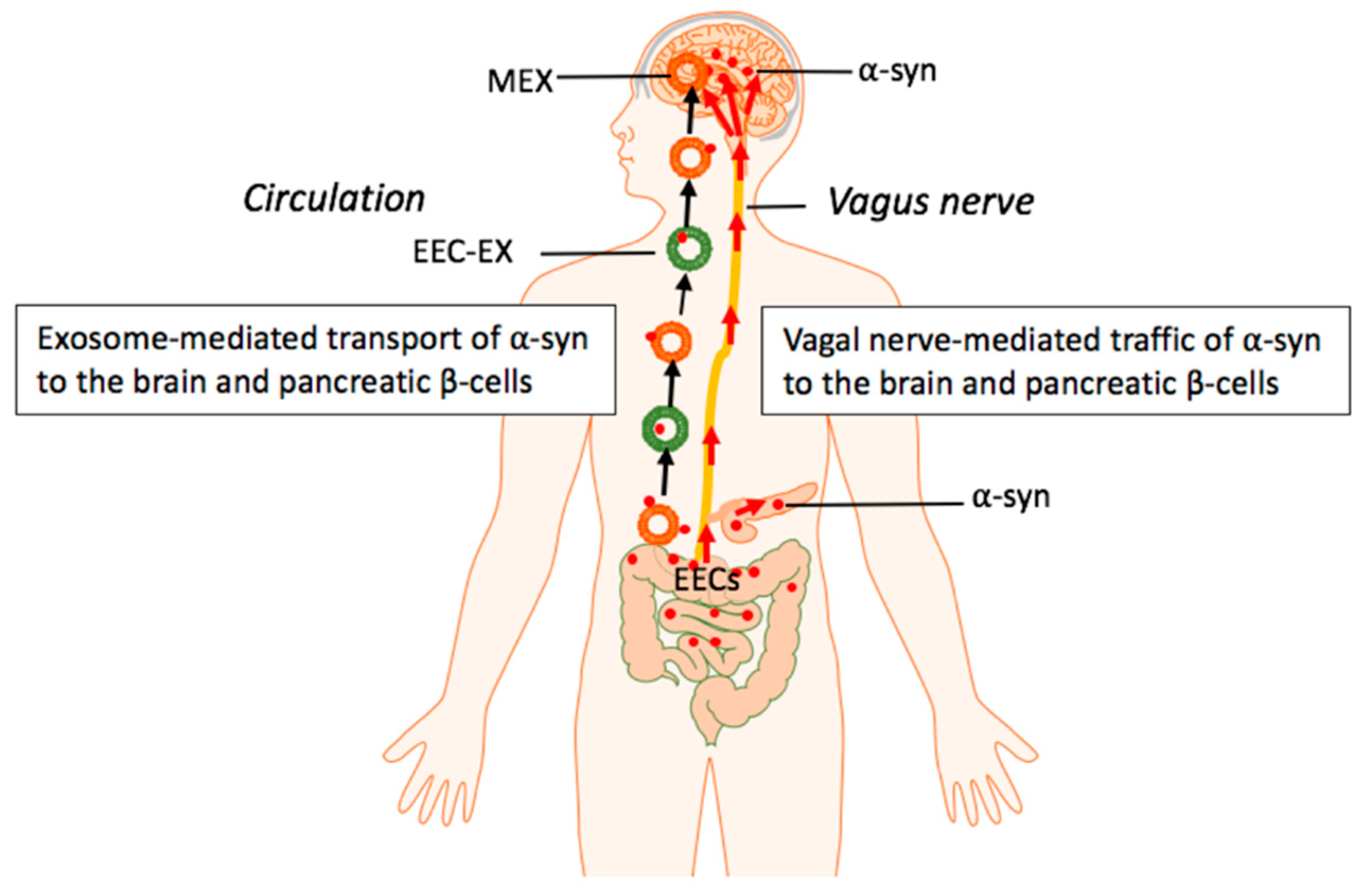{kind=link}
| Total Dairy | Milk | Cheese | Yogurt | References |
|---|---|---|---|---|
| + | + | Chen et al. (2002) [40] | ||
| + | Park et al. (2005) [41] | |||
| + | Sääksjärvi et al. (2013) [42] | |||
| + | Kyrozis et al. (2013) [43] | |||
| + | + | Jiang et al. (2014) [44] | ||
| + | Hughes et al. (2017) [45] | |||
| + | Olsson et al. (2020) [46] |
| Agent | Predicted Functions | References |
|---|---|---|
| MEX miRNA-148a | Suppression of DNMT1, hypomethylation of SNCA with increased expression of α-syn in EECs and SN neuron. | [114,125] |
| Suppression of AMPK activity increasing mTORC1 activity resulting in reduced ULK-1-mediated autophagy. | [206] | |
| Suppression of PGC-1α reducing the expression of LC3-II, SQSTM1 and LAMP2 resulting in reduced autophagy. | [288] | |
| Suppression of PGC-1α-mediated mitochondrial function with increased oxidative stress promoting α-syn aggregation. | [280,281,282,283,284,285] | |
| MEX miRNA-21 | Reduction of DNMT1 activity, hypomethylation of SNCA with increased expression of α-syn in EECs and SN neurons. | [126] |
| Suppression of LAMP2A resulting in reduced chaperone-mediated autophagy, accumulation of α-syn promoting α-syn aggregation. | [195,196,197] | |
| Exosome lipids | Promotion of α-syn aggregation. | [149,158] |
| Exosome membrane | α-syn binding to exosome outer membrane promoting exosome α-syn spreading. | [152] |
| Pathogenic Factors | PD | References | T2D | References |
|---|---|---|---|---|
| Exosome traffic | + | [149,150,151,156,158,227,340,341,342,343] | + | [294,295,296,297,298,299,344,345,346] |
| Vagal connectivity | + | [20,21,22,23,24,27,28,29] | + | [300,301,308,311] |
| α-Synuclein aggregation ↑ | + | [134,145,191] | + | [311] |
| Mitochondrial function ↓ | + | [13,179,184,221,225,228,262] | + | [328] |
| Oxidative stress ↑ | + | [222,223,237,238,242] | + | [326] |
| Autophagy ↓ | + | [94,154,177,178,183,185,188,189,190,191,192,196] | + | [327,328,329] |
| Increased mTORC1 ↑ | + | [82,83,84,85] | + | [57,58,266] |
| AMPK ↓ | + | [90] | + | [59,60] |
| MiRNA-148a ↑ | + | [173] | + | [206] |
| MiRNA-21 ↑ | + | [196] | + | [241,266] |
| BCAA ↑ | + | [263,265] | + | [268,269,270,271,272,273] |
| Metformin response | + | [89,90,91,92,93,265] | + | [61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnik, B.C. Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 1059. https://doi.org/10.3390/ijms22031059
Melnik BC. Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus. International Journal of Molecular Sciences. 2021; 22(3):1059. https://doi.org/10.3390/ijms22031059
Chicago/Turabian StyleMelnik, Bodo C. 2021. "Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus" International Journal of Molecular Sciences 22, no. 3: 1059. https://doi.org/10.3390/ijms22031059
APA StyleMelnik, B. C. (2021). Synergistic Effects of Milk-Derived Exosomes and Galactose on α-Synuclein Pathology in Parkinson’s Disease and Type 2 Diabetes Mellitus. International Journal of Molecular Sciences, 22(3), 1059. https://doi.org/10.3390/ijms22031059