
Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
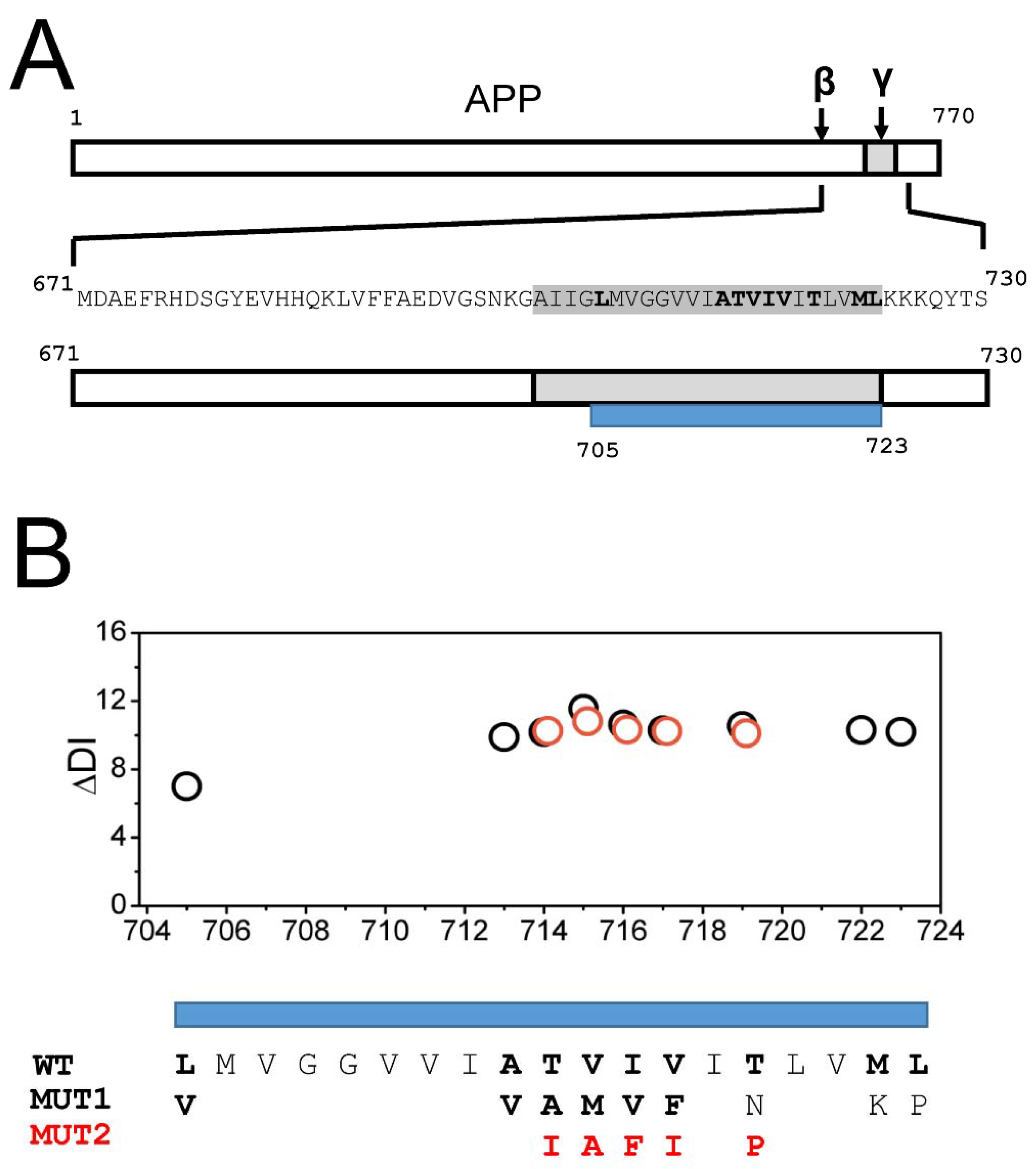
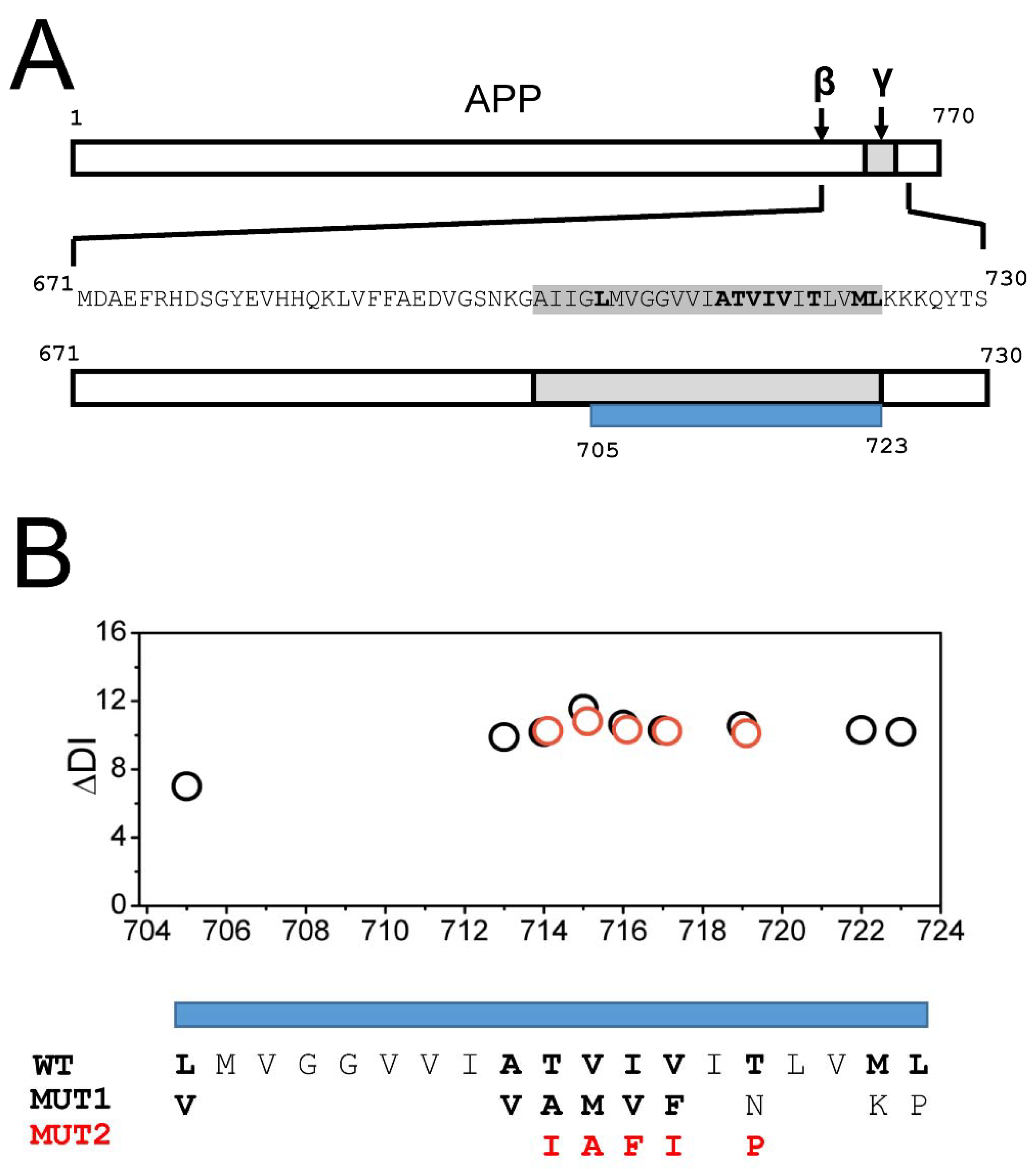
2.1. AD Mutations Destabilize the Secondary Structure of the APP Transmembrane Domain
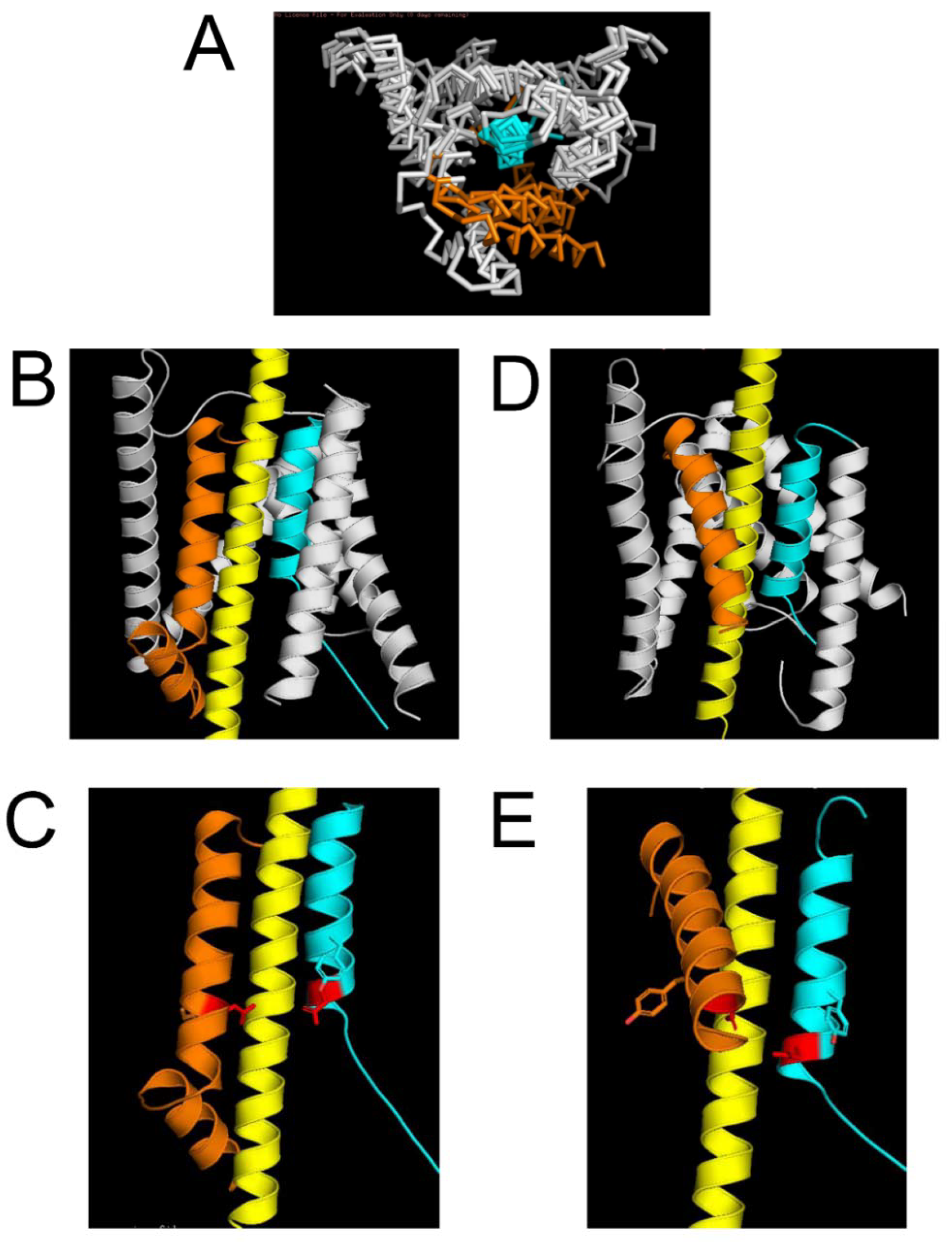
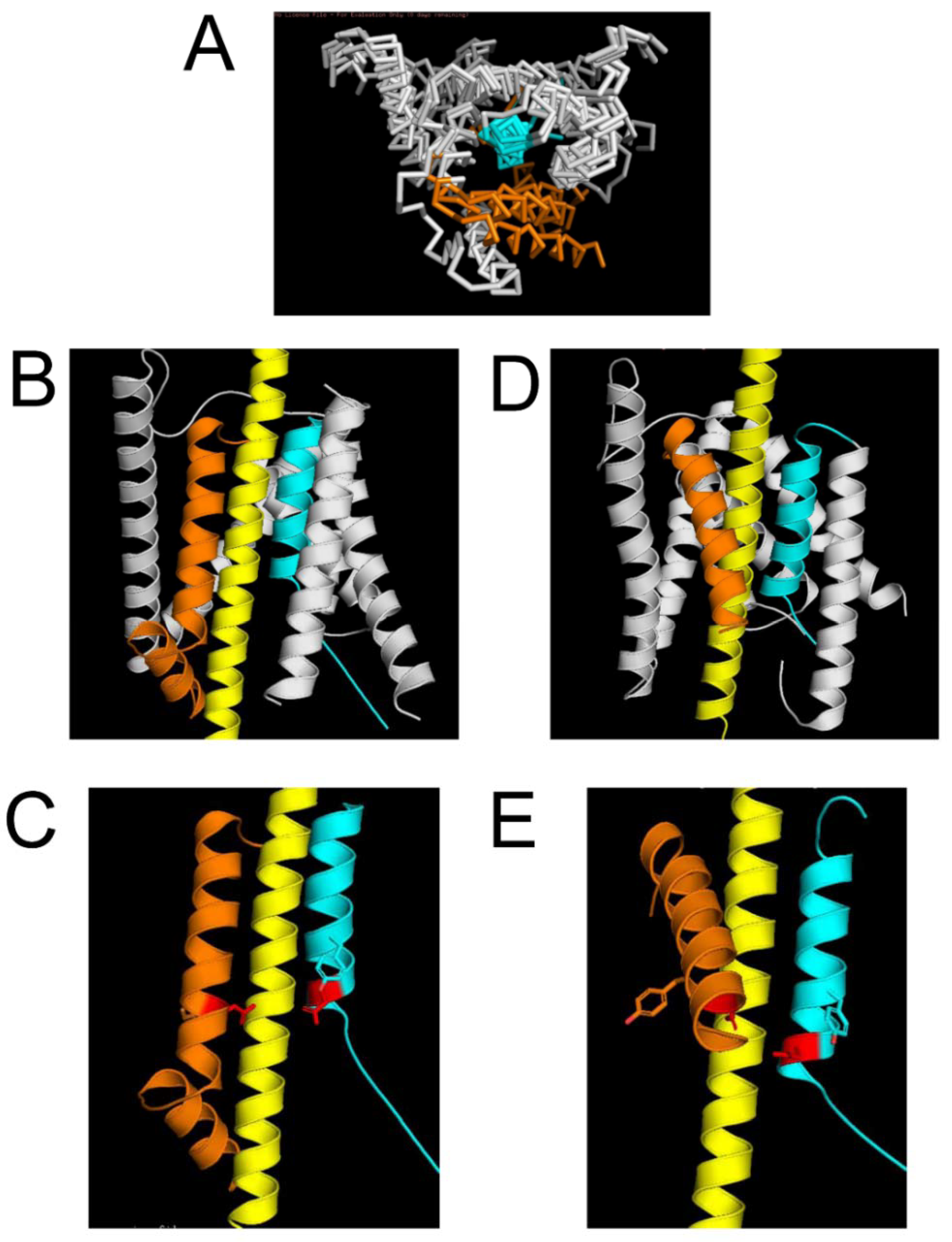
2.2. Protein Structural Models of the APP Complex with γ-Secretase
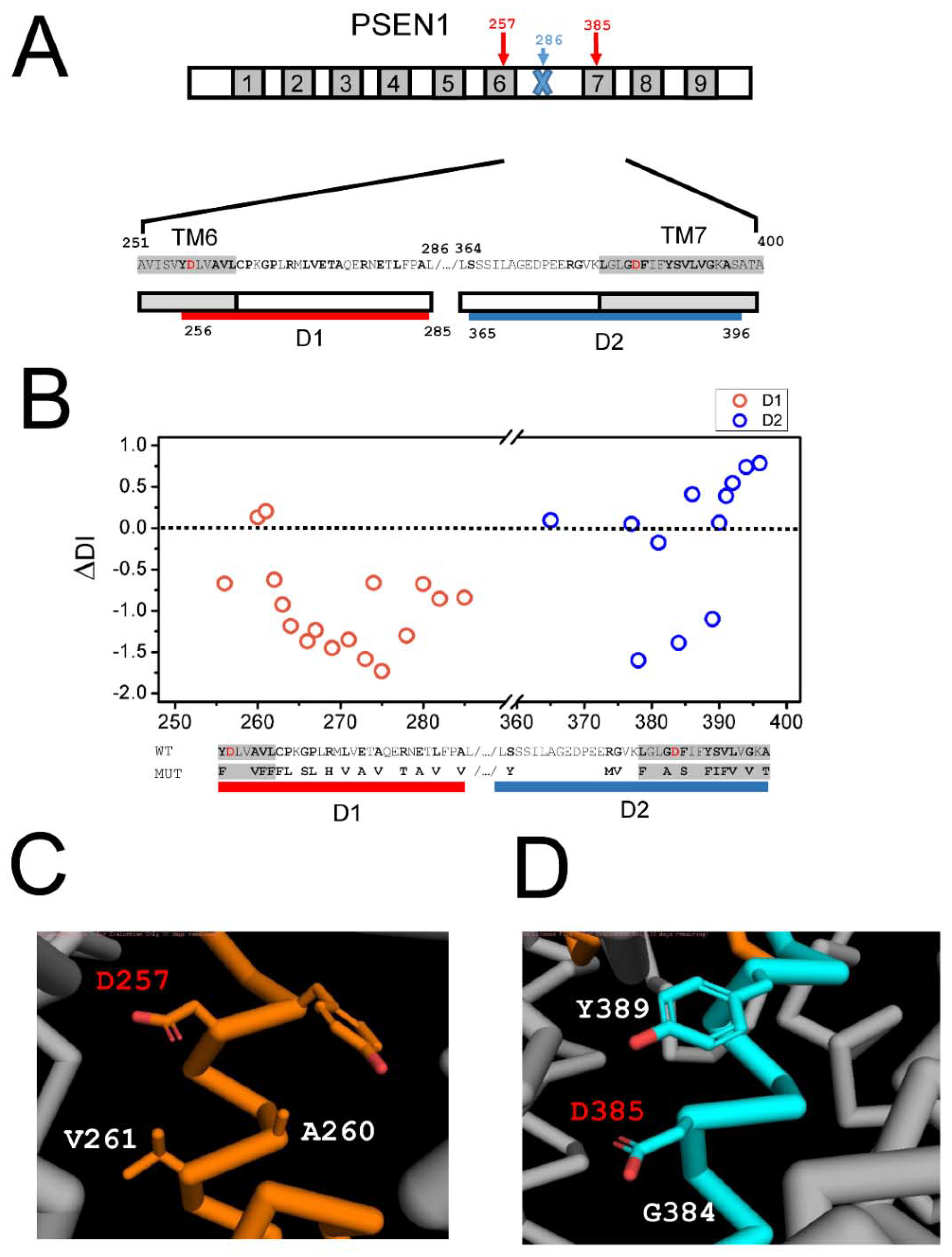
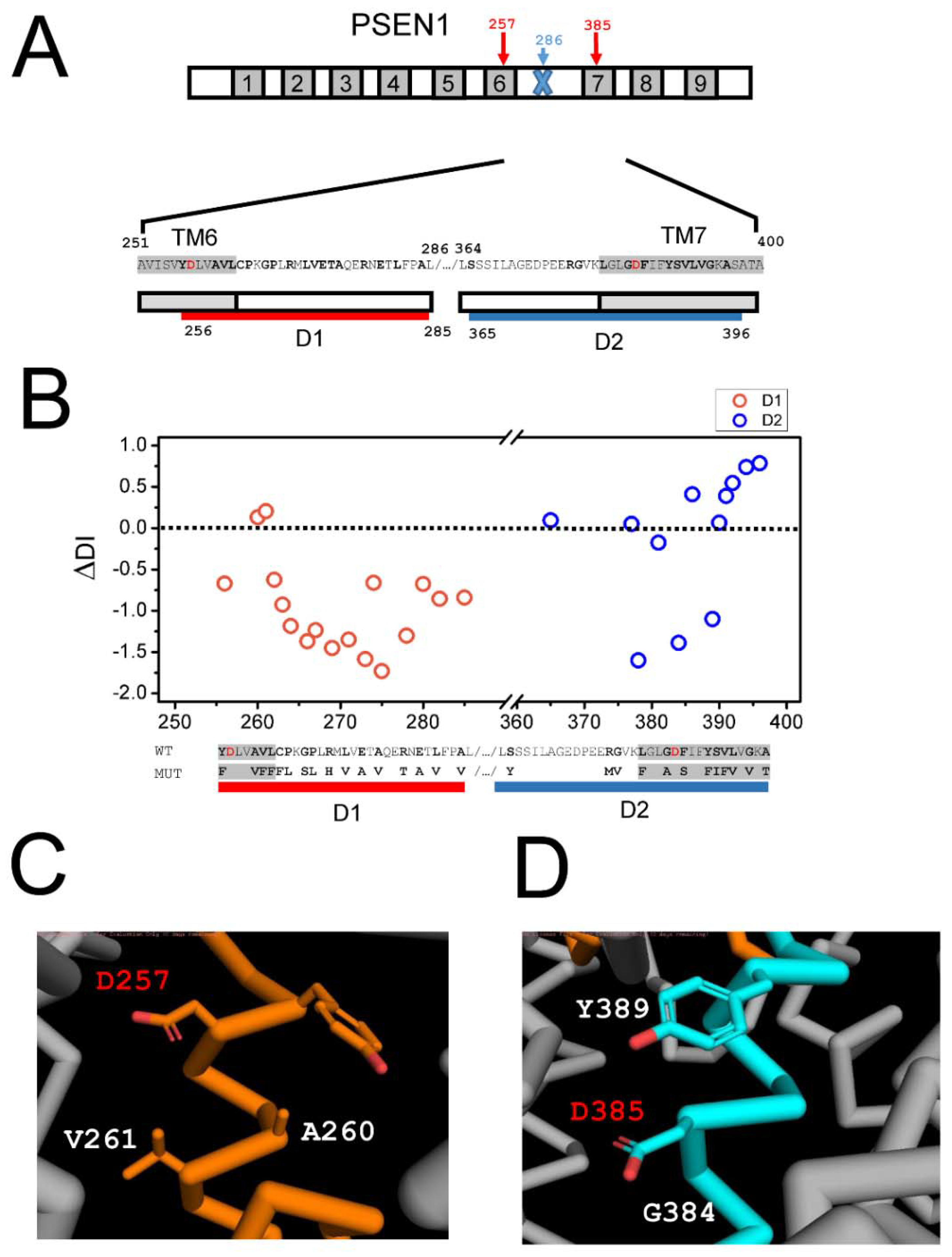
2.3. Effects of AD Mutations in PS1 on the Stability of the APP Complex with γ-Secretase
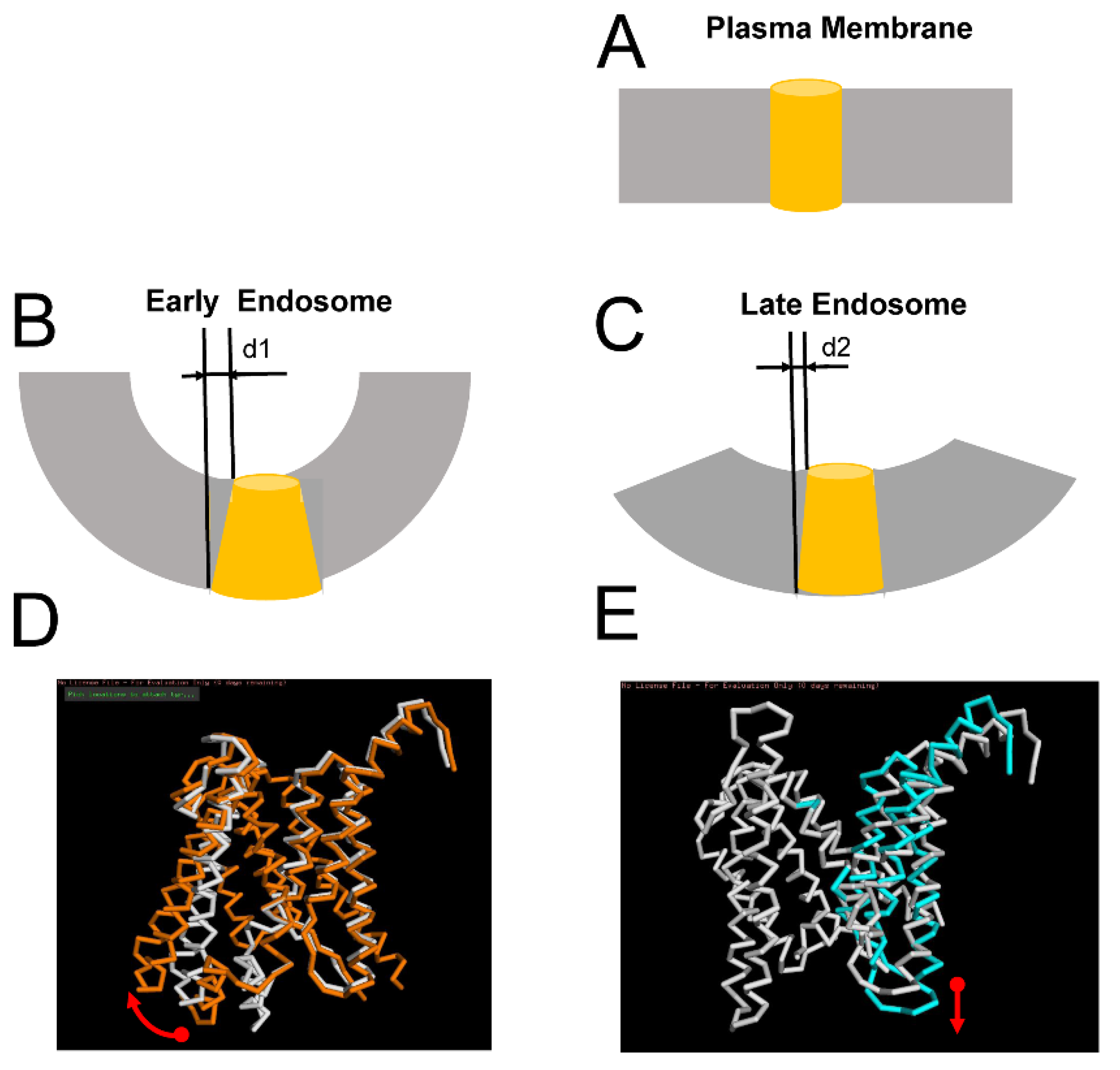
2.4. Effects of Membrane Curvature on APP Processing by γ-Secretase
3. Discussion
3.1. Comparison of M1 and M2 Models with Known Structures of the APP/γ-Secretase Complex
3.2. Potential Implications for the Development of Selective γ-Secretase Inhibitors
4. Materials and Methods
4.1. Calculations of the Disorder Index of FAD Mutations in APP and PS1
4.2. Structural Models of the APP Complex with γ-Secretase
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 1–23. [Google Scholar] [CrossRef]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Tolia, A.; De Strooper, B. Structure and function of gamma-secretase. Semin. Cell Dev. Biol. 2009, 20, 211–218. [Google Scholar] [CrossRef]
- Bergmans, B.A.; De Strooper, B. gamma-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. Gamma-secretase: Proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar] [CrossRef]
- De Strooper, B. Lessons from a failed gamma-secretase Alzheimer trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Albright, C.F.; Dockens, R.C.; Meredith, J.E., Jr.; Olson, R.E.; Slemmon, R.; Lentz, K.A.; Wang, J.S.; Denton, R.R.; Pilcher, G.; Rhyne, P.W.; et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J. Pharmacol. Exp. Ther. 2013, 344, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, S.C.; Kreft, A.F.; Harrison, B.; Abou-Gharbia, M.; Antane, M.; Aschmies, S.; Atchison, K.; Chlenov, M.; Cole, D.C.; Comery, T.; et al. Discovery of begacestat, a Notch-1-sparing gamma-secretase inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem. 2008, 51, 7348–7351. [Google Scholar] [CrossRef]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dockens, R.; Wang, J.S.; Castaneda, L.; Sverdlov, O.; Huang, S.P.; Slemmon, R.; Gu, H.; Wong, O.; Li, H.; Berman, R.M.; et al. A placebo-controlled, multiple ascending dose study to evaluate the safety, pharmacokinetics and pharmacodynamics of avagacestat (BMS-708163) in healthy young and elderly subjects. Clin. Pharmacokinet. 2012, 51, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.R. ACS chemical neuroscience molecule spotlight on Begacestat (GSI-953). ACS Chem. Neurosci. 2012, 3, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.J.; Castro, S.V.; Wang, F.; Pozdnyakov, N.; Ballard, T.E.; Sisodia, S.S.; Bales, K.R.; Johnson, D.S.; Li, Y.M. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 2012, 51, 7209–7211. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human gamma-secretase. eLife 2015, 4, e11182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, 6428. [Google Scholar] [CrossRef] [PubMed]
- Tcw, J.; Goate, A.M. Genetics of beta-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu, K.; Tomita, T. Membrane trafficking and proteolytic activity of gamma-secretase in Alzheimer’s disease. Biol. Chem. 2016, 397, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Ragvin, A.; Moro, E.; Fredman, D.; Navratilova, P.; Drivenes, O.; Engstrom, P.G.; Alonso, M.E.; de la Calle Mustienes, E.; Gomez Skarmeta, J.L.; Tavares, M.J.; et al. Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3. Proc. Natl. Acad. Sci. USA 2010, 107, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Ubelmann, F.; Burrinha, T.; Salavessa, L.; Gomes, R.; Ferreira, C.; Moreno, N.; Guimas Almeida, C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017, 18, 102–122. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.; Shelton, C.C.; Tian, Y.; Zhang, X.; Gilchrist, M.L.; Sisodia, S.S.; Li, Y.M. Activation and intrinsic gamma-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. USA 2010, 107, 21435–21440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedin-Weiss, S.; Caesar, I.; Winblad, B.; Blom, H.; Tjernberg, L.O. Super-resolution microscopy reveals gamma-secretase at both sides of the neuronal synapse. Acta Neuropathol. Commun. 2016, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/gamma-Secretase Determines Substrate Specificity and Generates an Intracellular Abeta Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2014, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Cooney, J.R.; Hurlburt, J.L.; Selig, D.K.; Harris, K.M.; Fiala, J.C. Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J. Neurosci. 2002, 22, 2215–2224. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Berezovska, O. Dynamic Nature of presenilin1/gamma-Secretase: Implication for Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2018, 55, 2275–2284. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Bezprozvanny, I. Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations. Int. J. Mol. Sci. 2021, 22, 13600. https://doi.org/10.3390/ijms222413600
Kim M, Bezprozvanny I. Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations. International Journal of Molecular Sciences. 2021; 22(24):13600. https://doi.org/10.3390/ijms222413600
Chicago/Turabian StyleKim, Meewhi, and Ilya Bezprozvanny. 2021. "Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations" International Journal of Molecular Sciences 22, no. 24: 13600. https://doi.org/10.3390/ijms222413600
APA StyleKim, M., & Bezprozvanny, I. (2021). Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations. International Journal of Molecular Sciences, 22(24), 13600. https://doi.org/10.3390/ijms222413600