
GSK3β Serine 389 Phosphorylation Modulates Cardiomyocyte Hypertrophy and Ischemic Injury

 ,
,  ,
,
Abstract
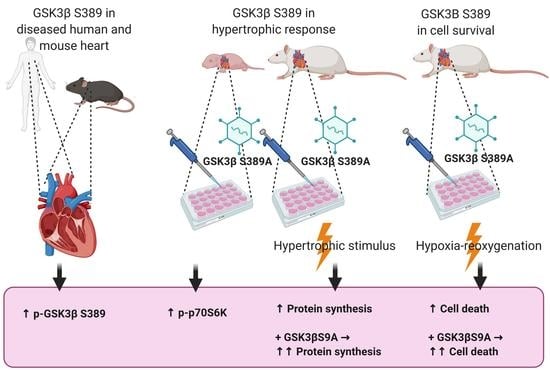
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
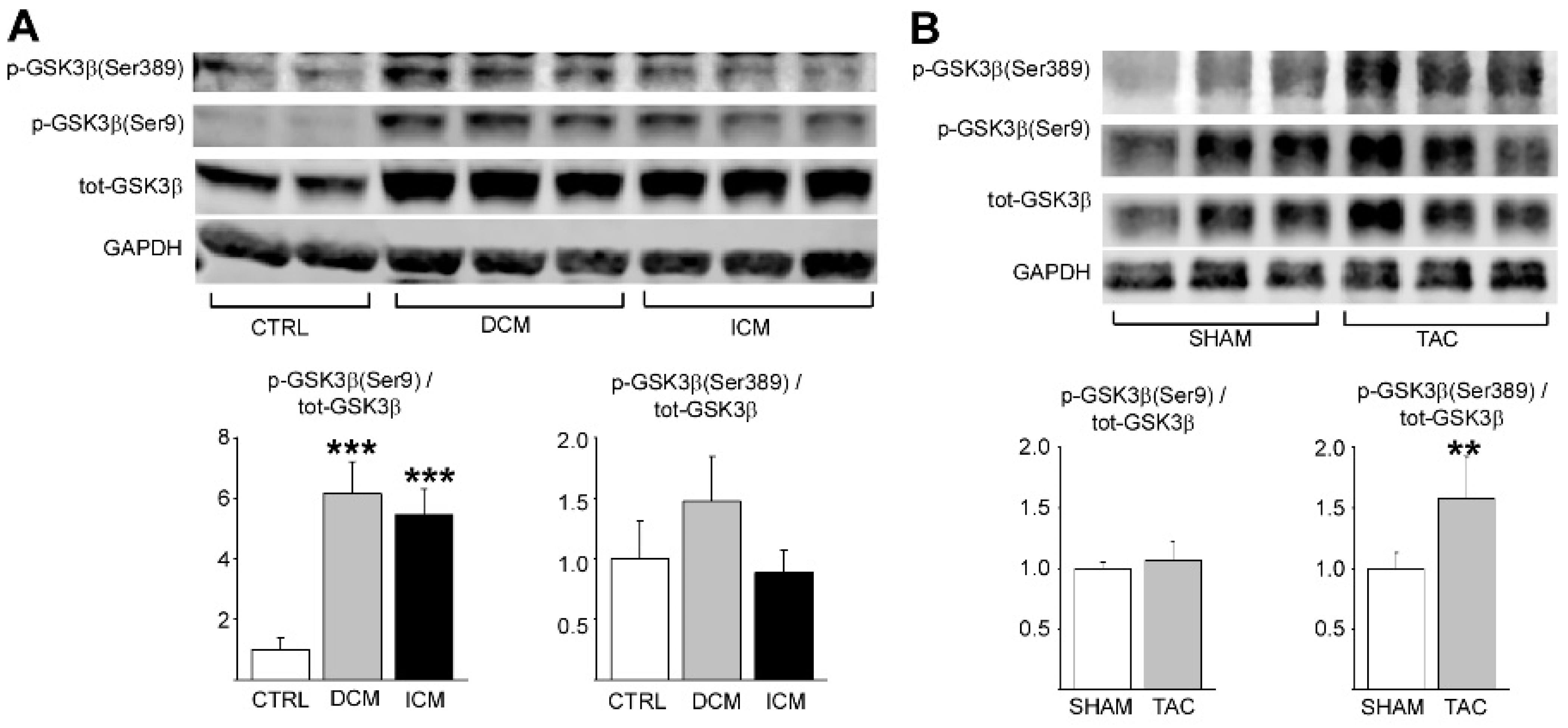
2.1. Phosphorylation of GSK3β S389 Is Increased in Diseased Human and Rodent Hearts
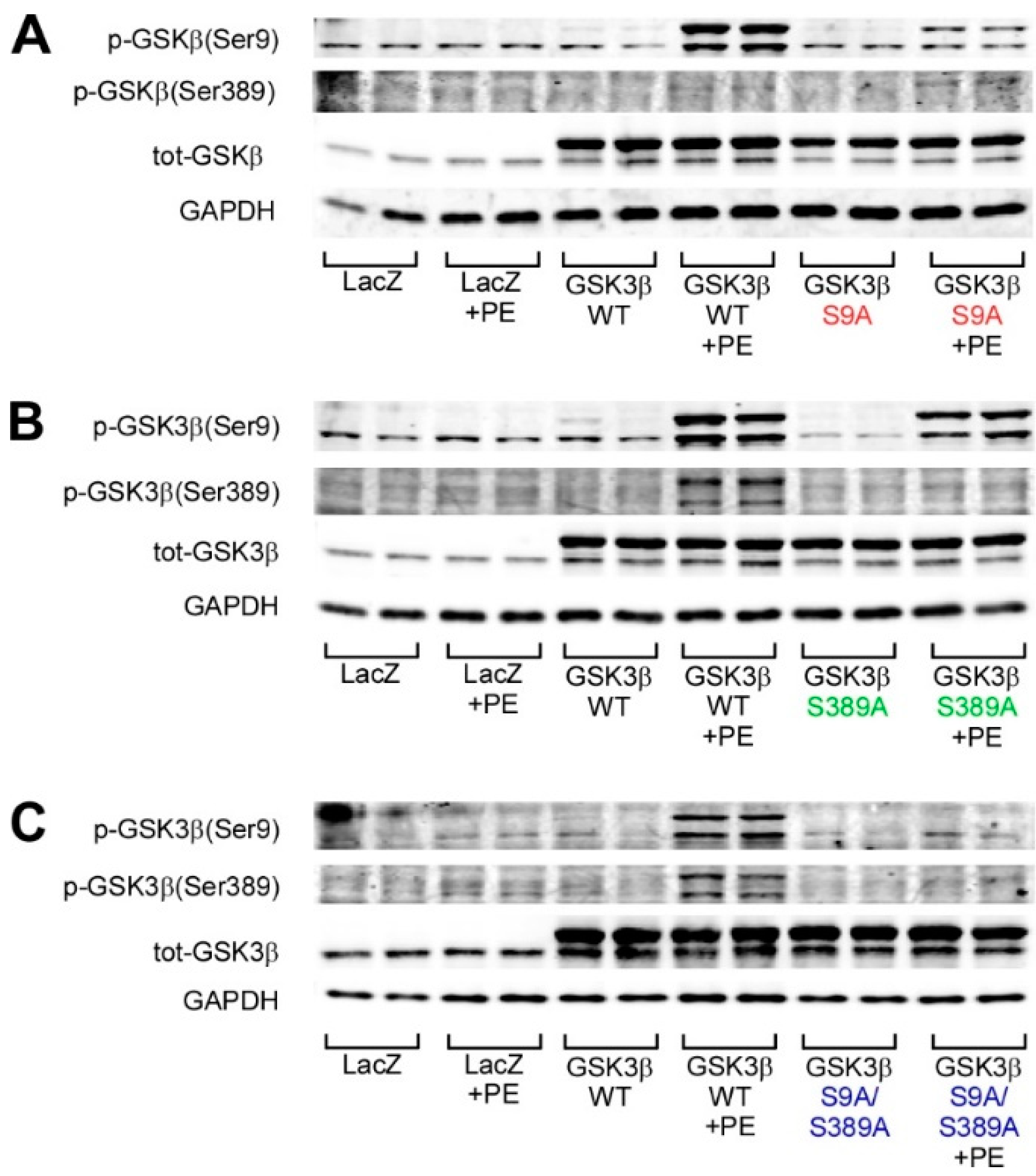
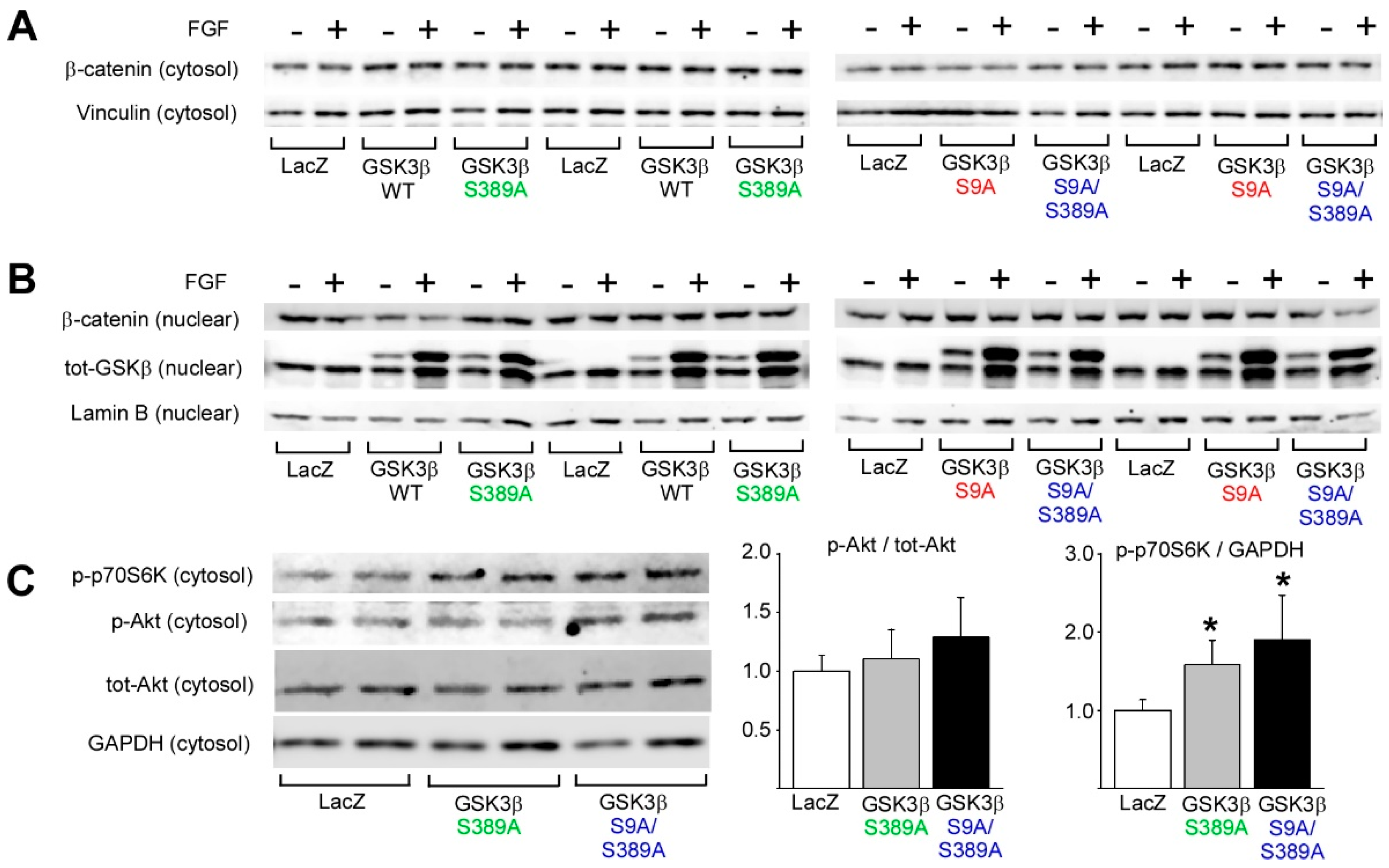
2.2. GSK3β S389 Phosphorylation Promotes GSK3β S9 Phosphorylation
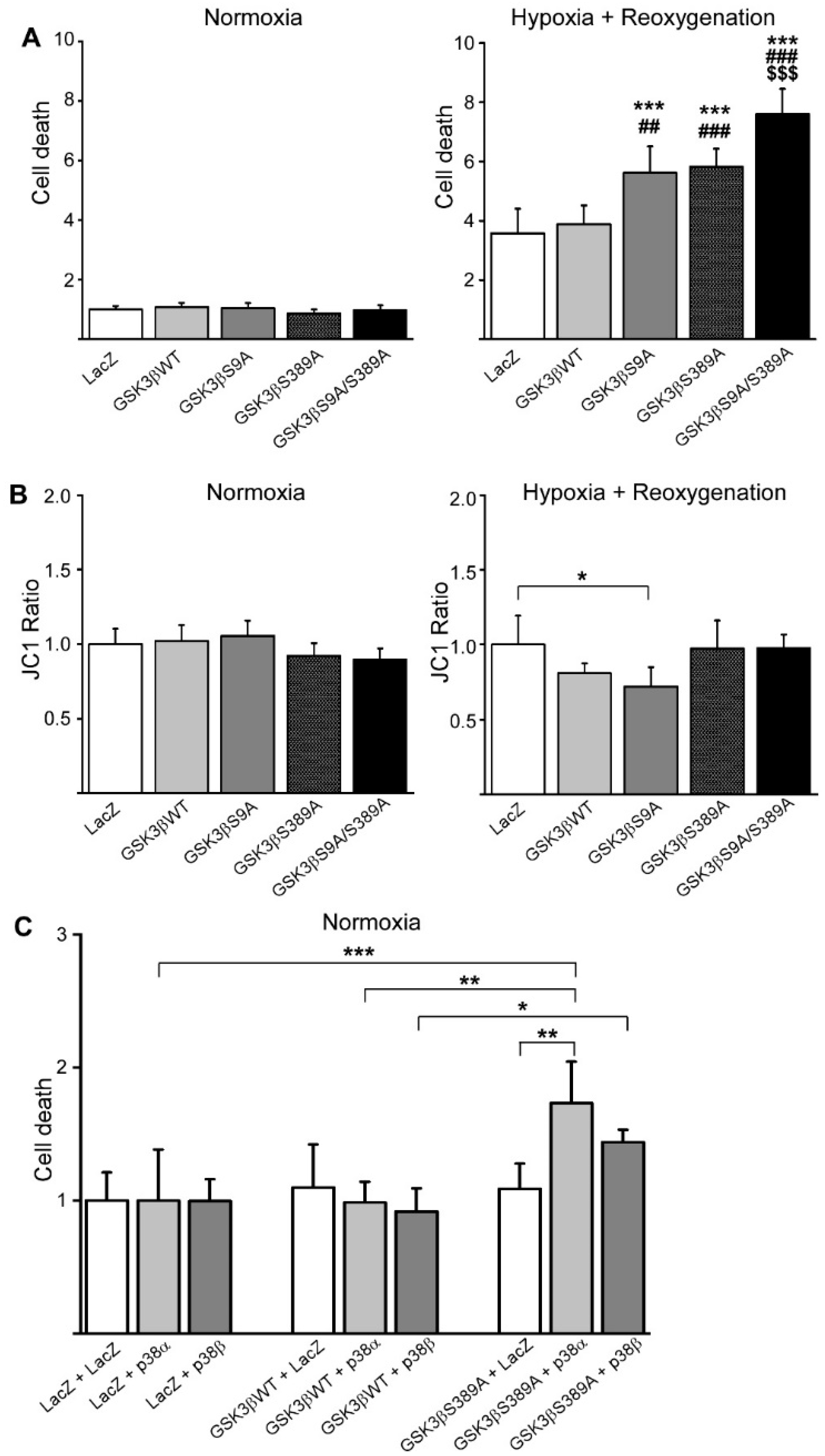
2.3. GSK3β S389 Regulates Cardiomyocyte Viability
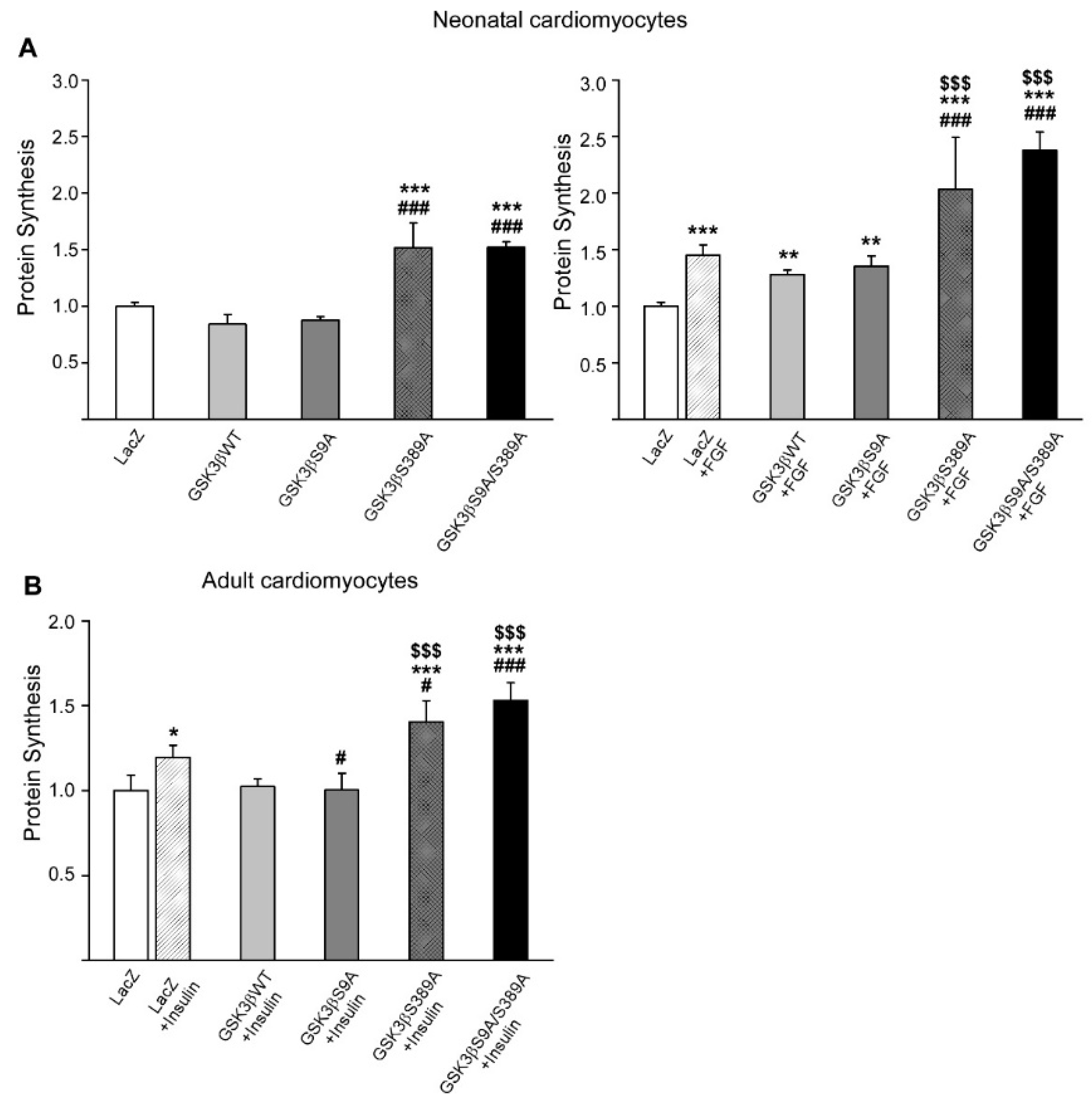
2.4. GSK3β S389 Regulates Cardiomyocyte Growth
2.5. GSK3β S389 Regulates mTORC1 Activity
3. Discussion
3.1. GSK3β S389 Phosphorylation Is Cardioprotective
3.2. GSK3β S389 Phosphorylation Attenuates Hypertrophic Cardiac Growth
3.3. GSK3β S389 Phosphorylation Attenuates Hypertrophy by Targeting mTORC1 Signaling Pathway
4. Materials and Methods
4.1. Human Cardiac Samples
4.2. Hemodynamic Pressure Overload
4.3. In Vitro Study Design
4.4. Recombinant Adenoviral Vectors
4.5. Isolation of Neonatal Rat Ventricular Cardiomyocytes
4.6. Isolation of Adult Rat Ventricular Cardiomyocytes
4.7. Adenoviral Infections
4.8. Measurement of Protein Synthesis
4.9. Hypoxia–Reoxygenation in Cell Culture
4.10. Cell Viability Assay
4.11. Western Blot Analysis
4.12. Mitochondrial Membrane Potential Assay
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report from the American Heart Association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef]
- GBD 2019 Diseases and Injuries Collaborators. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle. Separation from Cyclic-AMP-Dependent Protein Kinase and Phosphorylase Kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent Advances in Understanding the Cellular Roles of GSK-3. F1000Research 2017, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of Mutations in Wnt/Beta-Catenin, Hedgehog, Notch and PI3K Pathways on GSK-3 Activity-Diverse Effects on Cell Growth, Metabolism and Cancer. Biochim. Biophys. Acta 2016, 1863, 2942–2976. [Google Scholar] [CrossRef]
- Kerkela, R.; Woulfe, K.; Force, T. Glycogen Synthase Kinase-3β—Actively Inhibiting Hypertrophy. Trends Cardiovasc. Med. 2007, 17, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 Family as Therapeutic Target for Myocardial Diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C. What are the Bona Fide GSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic Discovery of in Vivo Phosphorylation Networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an Alternative Pathway for GSK3beta Inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, T.M.; Delgado, P.; Chen, L.; Salas, B.; Krementsov, D.; Fernandez, M.; Vernia, S.; Davis, R.J.; Heimann, R.; Teuscher, C.; et al. Inactivation of Nuclear GSK3beta by Ser(389) Phosphorylation Promotes Lymphocyte Fitness during DNA Double-Strand Break Response. Nat. Commun. 2016, 7, 10553. [Google Scholar] [CrossRef] [PubMed]
- Thornton, T.M.; Hare, B.; Colie, S.; Pendlebury, W.W.; Nebreda, A.R.; Falls, W.; Jaworski, D.M.; Rincon, M. Failure to Inactivate Nuclear GSK3beta by Ser(389)-Phosphorylation Leads to Focal Neuronal Death and Prolonged Fear Response. Neuropsychopharmacology 2018, 43, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Bonavaud, S.M.; Toole, B.J.; Yeaman, S.J. Regulation of Glycogen Synthesis by Amino Acids in Cultured Human Muscle Cells. J. Biol. Chem. 2001, 276, 952–956. [Google Scholar] [CrossRef] [Green Version]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3—At the Crossroads of Cell Death and Survival. J. Cell. Sci. 2014, 127, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR Signaling with Rapamycin Regresses Established Cardiac Hypertrophy Induced by Pressure Overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shende, P.; Plaisance, I.; Morandi, C.; Pellieux, C.; Berthonneche, C.; Zorzato, F.; Krishnan, J.; Lerch, R.; Hall, M.N.; Ruegg, M.A.; et al. Cardiac Raptor Ablation Impairs Adaptive Hypertrophy, Alters Metabolic Gene Expression, and Causes Heart Failure in Mice. Circulation 2011, 123, 1073–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 Regulates Cardiac Function and Myocyte Survival through 4E-BP1 Inhibition in Mice. J. Clin. Invest. 2010, 120, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian Target of Rapamycin Signaling in Cardiac Physiology and Disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin Attenuates Load-Induced Cardiac Hypertrophy in Mice. Circulation 2003, 107, 1664–1670. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Ferlito, M.; Aponte, A.; Kuno, A.; Miura, T.; Murphy, E.; Steenbergen, C. Pivotal Role of mTORC2 and Involvement of Ribosomal Protein S6 in Cardioprotective Signaling. Circ. Res. 2014, 114, 1268–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alakoski, T.; Ulvila, J.; Yrjola, R.; Vainio, L.; Magga, J.; Szabo, Z.; Licht, J.D.; Kerkela, R. Inhibition of Cardiomyocyte Sprouty1 Protects from Cardiac Ischemia-Reperfusion Injury. Basic Res. Cardiol. 2019, 114, 7. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.L.; Kumar, S.; Gao, F.; Louden, C.S.; Lopez, B.L.; Christopher, T.A.; Wang, C.; Lee, J.C.; Feuerstein, G.Z.; Yue, T.L. Inhibition of p38 Mitogen-Activated Protein Kinase Decreases Cardiomyocyte Apoptosis and Improves Cardiac Function after Myocardial Ischemia and Reperfusion. Circulation 1999, 99, 1685–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G. Molecular Basis of Cardioprotection: Signal Transduction in Ischemic Pre-, Post-, and Remote Conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef]
- Denise Martin, E.; De Nicola, G.F.; Marber, M.S. New Therapeutic Targets in Cardiology: p38 Alpha Mitogen-Activated Protein Kinase for Ischemic Heart Disease. Circulation 2012, 126, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of Beta-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wolgamott, L.; Yu, Y.; Blenis, J.; Yoon, S.O. Glycogen Synthase Kinase (GSK)-3 Promotes p70 Ribosomal Protein S6 Kinase (p70S6K) Activity and Cell Proliferation. Proc. Natl. Acad. Sci. USA. 2011, 108, E1204–E1213. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A.M. Crosstalks of GSK3 Signaling with the mTOR Network and Effects on Targeted Therapy of Cancer. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118635. [Google Scholar] [CrossRef] [Green Version]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen Synthase Kinase-3beta Mediates Convergence of Protection Signaling to Inhibit the Mitochondrial Permeability Transition Pore. J. Clin. Invest. 2004, 113, 1535–1549. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Tanno, M. Mitochondria and GSK-3beta in Cardioprotection Against Ischemia/Reperfusion Injury. Cardiovasc. Drugs Ther. 2010, 24, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Imahashi, K.; Steenbergen, C.; Murphy, E. Phosphorylation of Glycogen Synthase Kinase-3beta during Preconditioning through a Phosphatidylinositol-3-Kinase—Dependent Pathway is Cardioprotective. Circ. Res. 2002, 90, 377–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, L.; Paillard, M.; Thibault, H.; Derumeaux, G.; Ovize, M. Inhibition of GSK3beta by Postconditioning is Required to Prevent Opening of the Mitochondrial Permeability Transition Pore during Reperfusion. Circulation 2008, 117, 2761–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, Y.; Webb, I.G.; Davidson, S.M.; Ahmed, A.I.; Clark, J.E.; Jacquet, S.; Shah, A.M.; Miura, T.; Yellon, D.M.; Avkiran, M.; et al. Glycogen Synthase Kinase-3 Inactivation is Not Required for Ischemic Preconditioning or Postconditioning in the Mouse. Circ. Res. 2008, 103, 307–314. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, M.; Zhang, W.; Lu, X.Y. Adiponectin Stimulates Proliferation of Adult Hippocampal Neural Stem/Progenitor Cells through Activation of p38 Mitogen-Activated Protein Kinase (p38MAPK)/Glycogen Synthase Kinase 3beta (GSK-3beta)/Beta-Catenin Signaling Cascade. J. Biol. Chem. 2011, 286, 44913–44920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, I.G.; Sicard, P.; Clark, J.E.; Redwood, S.; Marber, M.S. Myocardial Stress Remodelling After Regional Infarction is Independent of Glycogen Synthase Kinase-3 Inactivation. J. Mol. Cell. Cardiol. 2010, 49, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Antos, C.L.; McKinsey, T.A.; Frey, N.; Kutschke, W.; McAnally, J.; Shelton, J.M.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Activated Glycogen Synthase-3 Beta Suppresses Cardiac Hypertrophy in Vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 907–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct Roles of GSK-3alpha and GSK-3beta Phosphorylation in the Heart Under Pressure Overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef] [Green Version]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen Synthase Kinase-3beta is a Negative Regulator of Cardiomyocyte Hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Huang, H.; Garcia Abreu, J.; He, X. Inhibition of GSK3 Phosphorylation of Beta-Catenin Via Phosphorylated PPPSPXS Motifs of Wnt Coreceptor LRP6. PLoS ONE 2009, 4, e4926. [Google Scholar] [CrossRef] [Green Version]
- Morisco, C.; Seta, K.; Hardt, S.E.; Lee, Y.; Vatner, S.F.; Sadoshima, J. Glycogen Synthase Kinase 3beta Regulates GATA4 in Cardiac Myocytes. J. Biol. Chem. 2001, 276, 28586–28597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, J.W.; Clipstone, N.A. Glycogen Synthase Kinase-3 Inhibits the DNA Binding Activity of NFATc. J. Biol. Chem. 2001, 276, 3666–3673. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls C-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatini, D.M. Twenty-Five Years of mTOR: Uncovering the Link from Nutrients to Growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, H.; Komuro, I.; Zou, Y.; Kudoh, S.; Yamazaki, T.; Yazaki, Y. Activation of p70 S6 Protein Kinase is Necessary for Angiotensin II-Induced Hypertrophy in Neonatal Rat Cardiac Myocytes. FEBS Lett. 1996, 379, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of Glycogen Synthase Kinase-3 Beta by Phosphorylation: New Kinase Connections in Insulin and Growth-Factor Signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Szabo, Z.; Magga, J.; Alakoski, T.; Ulvila, J.; Piuhola, J.; Vainio, L.; Kivirikko, K.I.; Vuolteenaho, O.; Ruskoaho, H.; Lipson, K.E.; et al. Connective Tissue Growth Factor Inhibition Attenuates Left Ventricular Remodeling and Dysfunction in Pressure Overload-Induced Heart Failure. Hypertension 2014, 63, 1235–1240. [Google Scholar] [CrossRef] [Green Version]
- Moilanen, A.M.; Rysa, J.; Serpi, R.; Mustonen, E.; Szabo, Z.; Aro, J.; Napankangas, J.; Tenhunen, O.; Sutinen, M.; Salo, T.; et al. (Pro)Renin Receptor Triggers Distinct Angiotensin II-Independent Extracellular Matrix Remodeling and Deterioration of Cardiac Function. PLoS ONE 2012, 7, e41404. [Google Scholar] [CrossRef]
- Koivisto, E.; Kaikkonen, L.; Tokola, H.; Pikkarainen, S.; Aro, J.; Pennanen, H.; Karvonen, T.; Rysa, J.; Kerkela, R.; Ruskoaho, H. Distinct Regulation of B-Type Natriuretic Peptide Transcription by p38 MAPK Isoforms. Mol. Cell. Endocrinol. 2011, 338, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, L.; Magga, J.; Ronkainen, V.P.; Koivisto, E.; Perjes, A.; Chuprun, J.K.; Vinge, L.E.; Kilpio, T.; Aro, J.; Ulvila, J.; et al. p38alpha Regulates SERCA2a Function. J. Mol. Cell. Cardiol. 2014, 67, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, J.S.; Raake, P.; Vinge, L.E.; DeGeorge, B.R., Jr.; Chuprun, J.K.; Harris, D.M.; Gao, E.; Eckhart, A.D.; Pitcher, J.A.; Koch, W.J. Uncovering G Protein-Coupled Receptor Kinase-5 as a Histone Deacetylase Kinase in the Nucleus of Cardiomyocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 12457–12462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivisto, E.; Karkkola, L.; Majalahti, T.; Aro, J.; Tokola, H.; Kerkela, R.; Ruskoaho, H. M-CAT Element Mediates Mechanical Stretch-Activated Transcription of B-Type Natriuretic Peptide Via ERK Activation. Can. J. Physiol. Pharmacol. 2011, 89, 539–550. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vainio, L.; Taponen, S.; Kinnunen, S.M.; Halmetoja, E.; Szabo, Z.; Alakoski, T.; Ulvila, J.; Junttila, J.; Lakkisto, P.; Magga, J.; et al. GSK3β Serine 389 Phosphorylation Modulates Cardiomyocyte Hypertrophy and Ischemic Injury. Int. J. Mol. Sci. 2021, 22, 13586. https://doi.org/10.3390/ijms222413586
Vainio L, Taponen S, Kinnunen SM, Halmetoja E, Szabo Z, Alakoski T, Ulvila J, Junttila J, Lakkisto P, Magga J, et al. GSK3β Serine 389 Phosphorylation Modulates Cardiomyocyte Hypertrophy and Ischemic Injury. International Journal of Molecular Sciences. 2021; 22(24):13586. https://doi.org/10.3390/ijms222413586
Chicago/Turabian StyleVainio, Laura, Saija Taponen, Sini M. Kinnunen, Eveliina Halmetoja, Zoltan Szabo, Tarja Alakoski, Johanna Ulvila, Juhani Junttila, Päivi Lakkisto, Johanna Magga, and et al. 2021. "GSK3β Serine 389 Phosphorylation Modulates Cardiomyocyte Hypertrophy and Ischemic Injury" International Journal of Molecular Sciences 22, no. 24: 13586. https://doi.org/10.3390/ijms222413586
APA StyleVainio, L., Taponen, S., Kinnunen, S. M., Halmetoja, E., Szabo, Z., Alakoski, T., Ulvila, J., Junttila, J., Lakkisto, P., Magga, J., & Kerkelä, R. (2021). GSK3β Serine 389 Phosphorylation Modulates Cardiomyocyte Hypertrophy and Ischemic Injury. International Journal of Molecular Sciences, 22(24), 13586. https://doi.org/10.3390/ijms222413586