
Delineation of the Ancestral Tus-Dependent Replication Fork Trap

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Ectopic Insertion of TerB, TerH and TerJ Sites
2.2. Chromosomal Binding of Tus
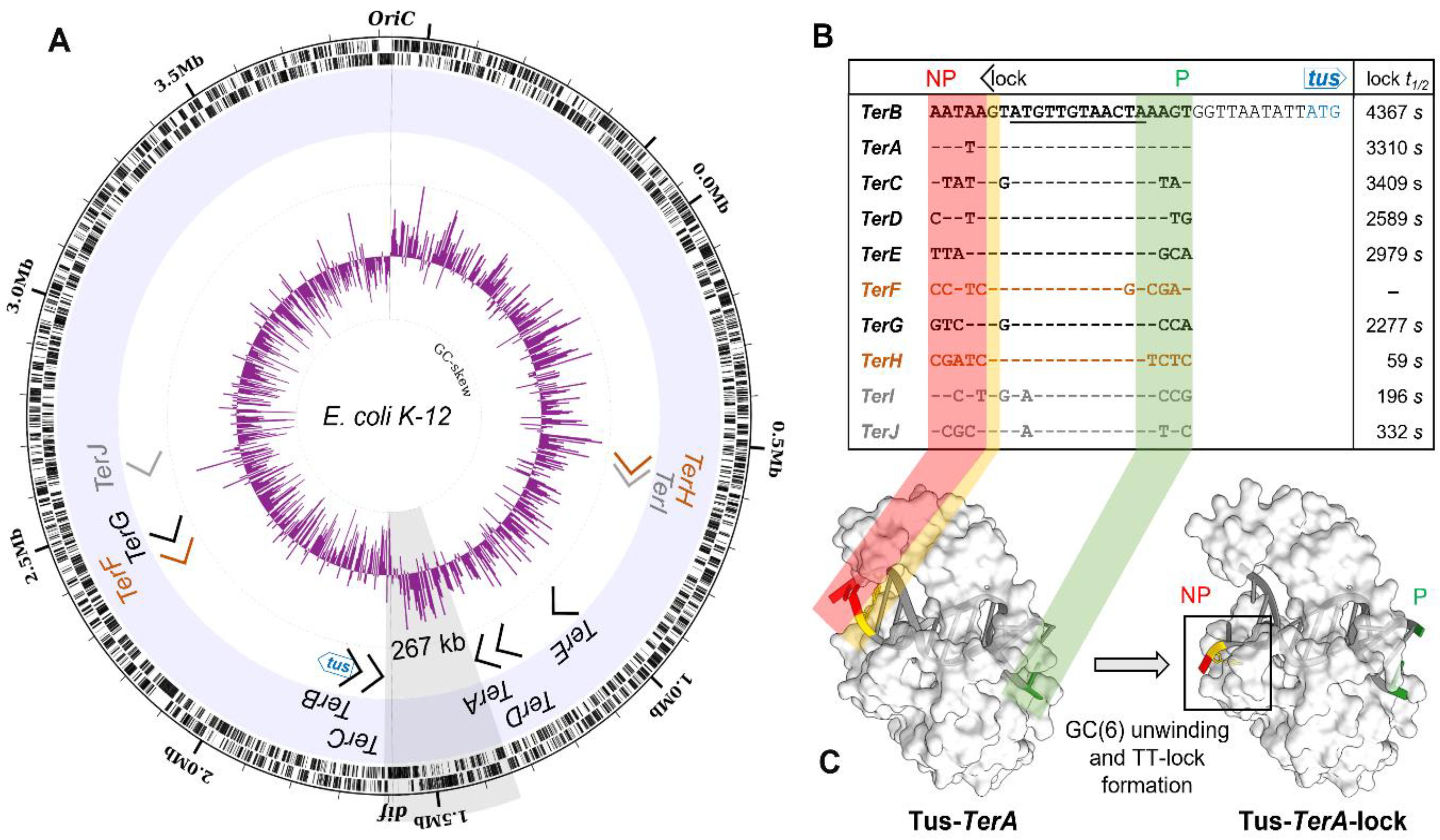
2.3. GC-Skew Relative to Termination Site Usage in E. coli
2.4. A Narrow Fork Trap Dyad in Edwardsiella Tarda
3. Discussion
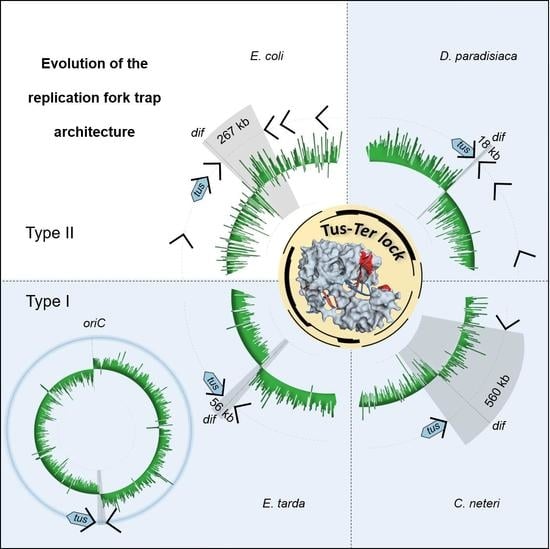
3.1. A Simplified Type II Replication Fork Trap in E. coli
3.2. The Ancestral Type I DNA Replication Fork Trap
3.3. Conclusions and Perspective
4. Materials and Methods
4.1. Plasmids and Protein Expression
4.2. Protein Expression and Crosslinking
4.3. Detection and Quantitation of GFP-Tagged Protein Expression
4.4. Chromosome Immunoprecipitation
4.5. qPCR Protocol
4.6. qPCR Analyses
4.7. Library Preparation and Illumina Sequencing
4.8. DNA Preparation for Nanopore Long Read Sequencing
4.9. Nanopore Long Read Sequencing and Genome Assembly
4.10. ChIP-Seq Analysis
4.11. Genome Engineering of Ectopic Ter Sites
4.12. Fork Trap Characterisation in Enterobacterales
4.13. Quantification and Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neylon, C.; Kralicek, A.V.; Hill, T.M.; Dixon, N.E. Replication termination in Escherichia coli: Structure and antihelicase activity of the Tus-Ter complex. Microbiol. Mol. Biol. Rev. 2005, 69, 501–526. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, B.A.; Raducanu, V.S.; Elshenawy, M.M.; Jergic, S.; Depken, M.; Dixon, N.E.; Hamdan, S.M.; Dekker, N.H. What is all this fuss about Tus? Comparison of recent findings from biophysical and biochemical experiments. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 49–63. [Google Scholar] [CrossRef]
- Xu, Z.Q.; Dixon, N.E. Bacterial replisomes. Curr. Opin. Struct. Biol. 2018, 53, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.J.; Schaeffer, P.M. A polyplex qPCR-based binding assay for protein-DNA interactions. Analyst 2012, 137, 4111–4113. [Google Scholar] [CrossRef]
- Moreau, M.J.; Schaeffer, P.M. Differential Tus-Ter binding and lock formation: Implications for DNA replication termination in Escherichia coli. Mol. Biosyst. 2012, 8, 2783–2791. [Google Scholar] [CrossRef]
- Duggin, I.G.; Bell, S.D. Termination structures in the Escherichia coli chromosome replication fork trap. J. Mol. Biol. 2009, 387, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Bastia, D.; Zzaman, S.; Krings, G.; Saxena, M.; Peng, X.; Greenberg, M.M. Replication termination mechanism as revealed by Tus-mediated polar arrest of a sliding helicase. Proc. Natl. Acad. Sci. USA 2008, 105, 12831–12836. [Google Scholar] [CrossRef]
- Kaplan, D.L. Replication termination: Mechanism of polar arrest revealed. Curr. Biol. 2006, 16, R684–R686. [Google Scholar] [CrossRef] [PubMed]
- Mulcair, M.D.; Schaeffer, P.M.; Oakley, A.J.; Cross, H.F.; Neylon, C.; Hill, T.M.; Dixon, N.E. A molecular mousetrap determines polarity of termination of DNA replication in E. coli. Cell 2006, 125, 1309–1319. [Google Scholar] [CrossRef]
- Neylon, C.; Brown, S.E.; Kralicek, A.V.; Miles, C.S.; Love, C.A.; Dixon, N.E. Interaction of the Escherichia coli replication terminator protein (Tus) with DNA: A model derived from DNA-binding studies of mutant proteins by surface plasmon resonance. Biochemistry 2000, 39, 11989–11999. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schaeffer, P.M.; Headlam, M.J.; Dixon, N.E. Protein—Protein interactions in the eubacterial replisome. IUBMB Life 2005, 57, 5–12. [Google Scholar] [CrossRef]
- Mulugu, S.; Potnis, A.; Shamsuzzaman; Taylor, J.; Alexander, K.; Bastia, D. Mechanism of termination of DNA replication of Escherichia coli involves helicase-contrahelicase interaction. Proc. Natl. Acad. Sci. USA 2001, 98, 9569–9574. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Elshenawy, M.M.; Jergic, S.; Takahashi, M.; Dixon, N.E.; Hamdan, S.M.; Patel, S.S. Two mechanisms coordinate replication termination by the Escherichia coli Tus-Ter complex. Nucl. Acids Res. 2015, 43, 5924–5935. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berghuis, B.A.; Dulin, D.; Xu, Z.Q.; van Laar, T.; Cross, B.; Janissen, R.; Jergic, S.; Dixon, N.E.; Depken, M.; Dekker, N.H. Strand separation establishes a sustained lock at the Tus-Ter replication fork barrier. Nat. Chem. Biol. 2015, 11, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Elshenawy, M.M.; Jergic, S.; Xu, Z.Q.; Sobhy, M.A.; Takahashi, M.; Oakley, A.J.; Dixon, N.E.; Hamdan, S.M. Replisome speed determines the efficiency of the Tus-Ter replication termination barrier. Nature 2015, 525, 394–398. [Google Scholar] [CrossRef]
- Kamada, K.; Horiuchi, T.; Ohsumi, K.; Shimamoto, N.; Morikawa, K. Structure of a replication-terminator protein complexed with DNA. Nature 1996, 383, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.J.; Schaeffer, P.M. Dissecting the salt dependence of the Tus-Ter protein-DNA complexes by high-throughput differential scanning fluorimetry of a GFP-tagged Tus. Mol. Biosyst. 2013, 9, 3146–3154. [Google Scholar] [CrossRef][Green Version]
- Coskun-Ari, F.F.; Hill, T.M. Sequence-specific interactions in the Tus-Ter complex and the effect of base pair substitutions on arrest of DNA replication in Escherichia coli. J. Biol. Chem. 1997, 272, 26448–26456. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Nishitani, H.; Kobayashi, T. A new type of E. coli recombinational hotspot which requires for activity both DNA replication termination events and the Chi sequence. Adv. Biophys. 1995, 31, 133–147. [Google Scholar] [CrossRef]
- Mohanty, B.K.; Bairwa, N.K.; Bastia, D. Contrasting roles of checkpoint proteins as recombination modulators at Fob1-Ter complexes with or without fork arrest. Eukaryot. Cell 2009, 8, 487–495. [Google Scholar] [CrossRef]
- Rothstein, R.; Michel, B.; Gangloff, S. Replication fork pausing and recombination or “gimme a break”. Genes Dev. 2000, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moolman, M.C.; Tiruvadi Krishnan, S.; Kerssemakers, J.W.; de Leeuw, R.; Lorent, V.; Sherratt, D.J.; Dekker, N.H. The progression of replication forks at natural replication barriers in live bacteria. Nucl. Acids Res. 2016, 44, 6262–6273. [Google Scholar] [CrossRef]
- Roecklein, B.; Pelletier, A.; Kuempel, P. The tus gene of Escherichia coli: Autoregulation, analysis of flanking sequences and identification of a complementary system in Salmonella typhimurium. Res. Microbiol. 1991, 142, 169–175. [Google Scholar] [CrossRef]
- Roecklein, B.A.; Kuempel, P.L. In vivo characterization of tus gene expression in Escherichia coli. Mol. Microbiol. 1992, 6, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Enyeart, P.J.; Chirieleison, S.M.; Dao, M.N.; Perutka, J.; Quandt, E.M.; Yao, J.; Whitt, J.T.; Keatinge-Clay, A.T.; Lambowitz, A.M.; Ellington, A.D. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol. Syst. Biol. 2013, 9, 685. [Google Scholar] [CrossRef]
- Bidnenko, V.; Ehrlich, S.D.; Michel, B. Replication fork collapse at replication terminator sequences. EMBO J. 2002, 21, 3898–3907. [Google Scholar] [CrossRef]
- Sharma, B.; Hill, T.M. Insertion of inverted Ter sites into the terminus region of the Escherichia coli chromosome delays completion of DNA replication and disrupts the cell cycle. Mol. Microbiol. 1995, 18, 45–61. [Google Scholar] [CrossRef]
- Bidnenko, V.; Lestini, R.; Michel, B. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol. Microbiol. 2006, 62, 382–396. [Google Scholar] [CrossRef]
- Esnault, E.; Valens, M.; Espeli, O.; Boccard, F. Chromosome structuring limits genome plasticity in Escherichia coli. PLoS Genet. 2007, 3, e226. [Google Scholar] [CrossRef]
- Poteete, A.R. What makes the bacteriophage lambda Red system useful for genetic engineering: Molecular mechanism and biological function. FEMS Microbiol. Lett. 2001, 201, 9–14. [Google Scholar] [PubMed]
- Mei, Q.; Fitzgerald, D.M.; Liu, J.; Xia, J.; Pribis, J.P.; Zhai, Y.; Nehring, R.B.; Paiano, J.; Li, H.; Nussenzweig, A.; et al. Two mechanisms of chromosome fragility at replication-termination sites in bacteria. Sci. Adv. 2021, 7, eabe2846. [Google Scholar] [CrossRef]
- Moreau, M.J.J.; Morin, I.; Askin, S.P.; Cooper, A.; Moreland, N.J.; Vasudevan, S.G.; Schaeffer, P.M. Rapid determination of protein stability and ligand binding by differential scanning fluorimetry of GFP-tagged proteins. RSC Adv. 2012, 2, 11892–11900. [Google Scholar] [CrossRef]
- Worning, P.; Jensen, L.J.; Hallin, P.F.; Staerfeldt, H.H.; Ussery, D.W. Origin of replication in circular prokaryotic chromosomes. Environ. Microbiol. 2006, 8, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Rocha, E.P. From GC skews to wavelets: A gentle guide to the analysis of compositional asymmetries in genomic data. Biochimie 2008, 90, 648–659. [Google Scholar] [CrossRef]
- Arakawa, K.; Tomita, M. The GC skew index: A measure of genomic compositional asymmetry and the degree of replicational selection. Evol. Bioinform. 2007, 3, 159–168. [Google Scholar] [CrossRef]
- Hendrickson, H.; Lawrence, J.G. Mutational bias suggests that replication termination occurs near the dif site, not at Ter sites. Mol. Microbiol. 2007, 64, 42–56. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K.; Tomita, M. Validation of bacterial replication termination models using simulation of genomic mutations. PLoS ONE 2012, 7, e34526. [Google Scholar]
- Kono, N.; Tomita, M.; Arakawa, K. Accelerated Laboratory Evolution Reveals the Influence of Replication on the GC Skew in Escherichia coli. Genome Biol. Evol. 2018, 10, 3110–3117. [Google Scholar] [PubMed]
- Galli, E.; Ferat, J.L.; Desfontaines, J.M.; Val, M.E.; Skovgaard, O.; Barre, F.X.; Possoz, C. Replication termination without a replication fork trap. Sci. Rep. 2019, 9, 8315. [Google Scholar] [CrossRef]
- Henderson, T.A.; Nilles, A.F.; Valjavec-Gratian, M.; Hill, T.M. Site-directed mutagenesis and phylogenetic comparisons of the Escherichia coli Tus protein: DNA-protein interactions alone can not account for Tus activity. Mol. Genet. Genom. 2001, 265, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.A.; Chandramouly, G.; Huang, B.; Kwok, A.; Follonier, C.; Deng, C.; Scully, R. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 2014, 510, 556–559. [Google Scholar] [CrossRef]
- Larsen, N.B.; Sass, E.; Suski, C.; Mankouri, H.W.; Hickson, I.D. The Escherichia coli Tus-Ter replication fork barrier causes site-specific DNA replication perturbation in yeast. Nat. Commun. 2014, 5, 3574. [Google Scholar] [CrossRef] [PubMed]
- Morin, I.; Dixon, N.E.; Schaeffer, P.M. Ultrasensitive detection of antibodies using a new Tus-Ter-lock immunoPCR system. Mol. Biosyst. 2010, 6, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Askin, S.P.; Schaeffer, P.M. A universal immuno-PCR platform for comparative and ultrasensitive quantification of dual affinity-tagged proteins in complex matrices. Analyst 2012, 137, 5193–5196. [Google Scholar] [CrossRef]
- Natarajan, S.; Kaul, S.; Miron, A.; Bastia, D. A 27 kd protein of E. coli promotes antitermination of replication in vitro at a sequence-specific replication terminus. Cell 1993, 72, 113–120. [Google Scholar] [CrossRef]
- Goodall, D.J.; Jameson, K.H.; Hawkins, M.; Rudolph, C.J. A Fork Trap in the Chromosomal Termination Area Is Highly Conserved across All Escherichia coli Phylogenetic Groups. Int. J. Mol. Sci. 2021, 22, 7928. [Google Scholar] [CrossRef] [PubMed]
- Dahdah, D.B.; Morin, I.; Moreau, M.J.; Dixon, N.E.; Schaeffer, P.M. Site-specific covalent attachment of DNA to proteins using a photoactivatable Tus-Ter complex. Chem. Commun. 2009, 3050–3052. [Google Scholar] [CrossRef]
- Moreau, M.J.; Morin, I.; Schaeffer, P.M. Quantitative determination of protein stability and ligand binding using a green fluorescent protein reporter system. Mol. Biosyst. 2010, 6, 1285–1292. [Google Scholar] [CrossRef]
- Morin, I.; Schaeffer, P.M.; Askin, S.P.; Dixon, N.E. Combining RNA-DNA swapping and quantitative polymerase chain reaction for the detection of influenza A nucleoprotein. Anal. Biochem. 2012, 420, 121–126. [Google Scholar]
- Volkmer, B.; Heinemann, M. Condition-dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS ONE 2011, 6, e23126. [Google Scholar] [CrossRef]
- Meijerink, J.; Mandigers, C.; van de Locht, L.; Tonnissen, E.; Goodsaid, F.; Raemaekers, J. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 2001, 3, 55–61. [Google Scholar] [CrossRef][Green Version]
- Isaacs, F.J.; Carr, P.A.; Wang, H.H.; Lajoie, M.J.; Sterling, B.; Kraal, L.; Tolonen, A.C.; Gianoulis, T.A.; Goodman, D.B.; Reppas, N.B.; et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 2011, 333, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain BL21(DE3) | Doubling Time TD in min (SE) |
|---|---|
| TerB (P) | 25.5 (0.4) |
| TerH (NP) | 24.1 (0.1) * |
| TerH (P) | 23.7 (0.6) |
| TerJ (P) | 22.7 (0.3) |
| TerJ (NP) | 23.7 (0.3) |
| Control | 25.9 (0.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toft, C.J.; Moreau, M.J.J.; Perutka, J.; Mandapati, S.; Enyeart, P.; Sorenson, A.E.; Ellington, A.D.; Schaeffer, P.M. Delineation of the Ancestral Tus-Dependent Replication Fork Trap. Int. J. Mol. Sci. 2021, 22, 13533. https://doi.org/10.3390/ijms222413533
Toft CJ, Moreau MJJ, Perutka J, Mandapati S, Enyeart P, Sorenson AE, Ellington AD, Schaeffer PM. Delineation of the Ancestral Tus-Dependent Replication Fork Trap. International Journal of Molecular Sciences. 2021; 22(24):13533. https://doi.org/10.3390/ijms222413533
Chicago/Turabian StyleToft, Casey J., Morgane J. J. Moreau, Jiri Perutka, Savitri Mandapati, Peter Enyeart, Alanna E. Sorenson, Andrew D. Ellington, and Patrick M. Schaeffer. 2021. "Delineation of the Ancestral Tus-Dependent Replication Fork Trap" International Journal of Molecular Sciences 22, no. 24: 13533. https://doi.org/10.3390/ijms222413533
APA StyleToft, C. J., Moreau, M. J. J., Perutka, J., Mandapati, S., Enyeart, P., Sorenson, A. E., Ellington, A. D., & Schaeffer, P. M. (2021). Delineation of the Ancestral Tus-Dependent Replication Fork Trap. International Journal of Molecular Sciences, 22(24), 13533. https://doi.org/10.3390/ijms222413533