
Generation of Induced Nephron Progenitor-like Cells from Human Urine-Derived Cells

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
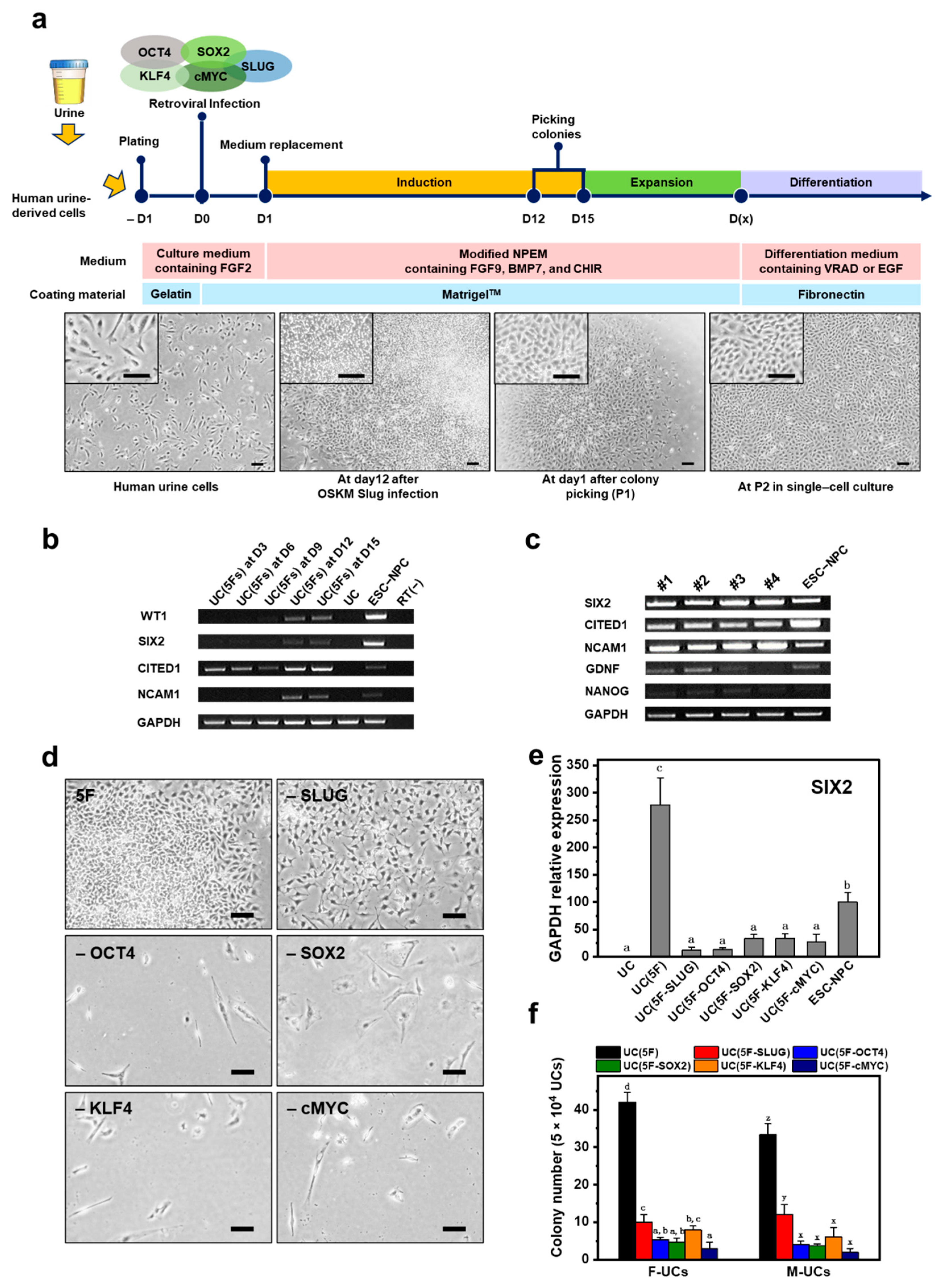
2.1. Screening for NPC-Inducing Factors
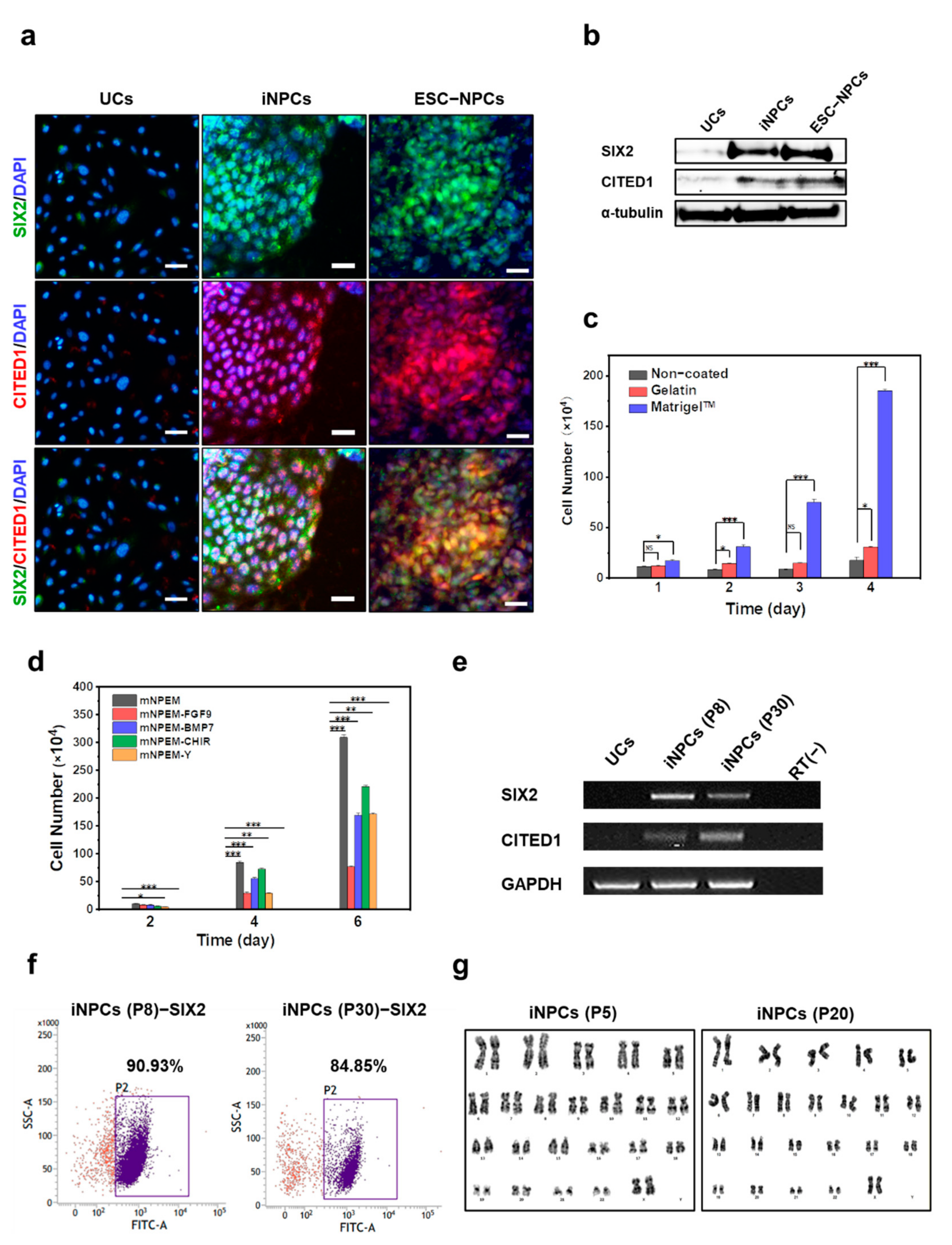
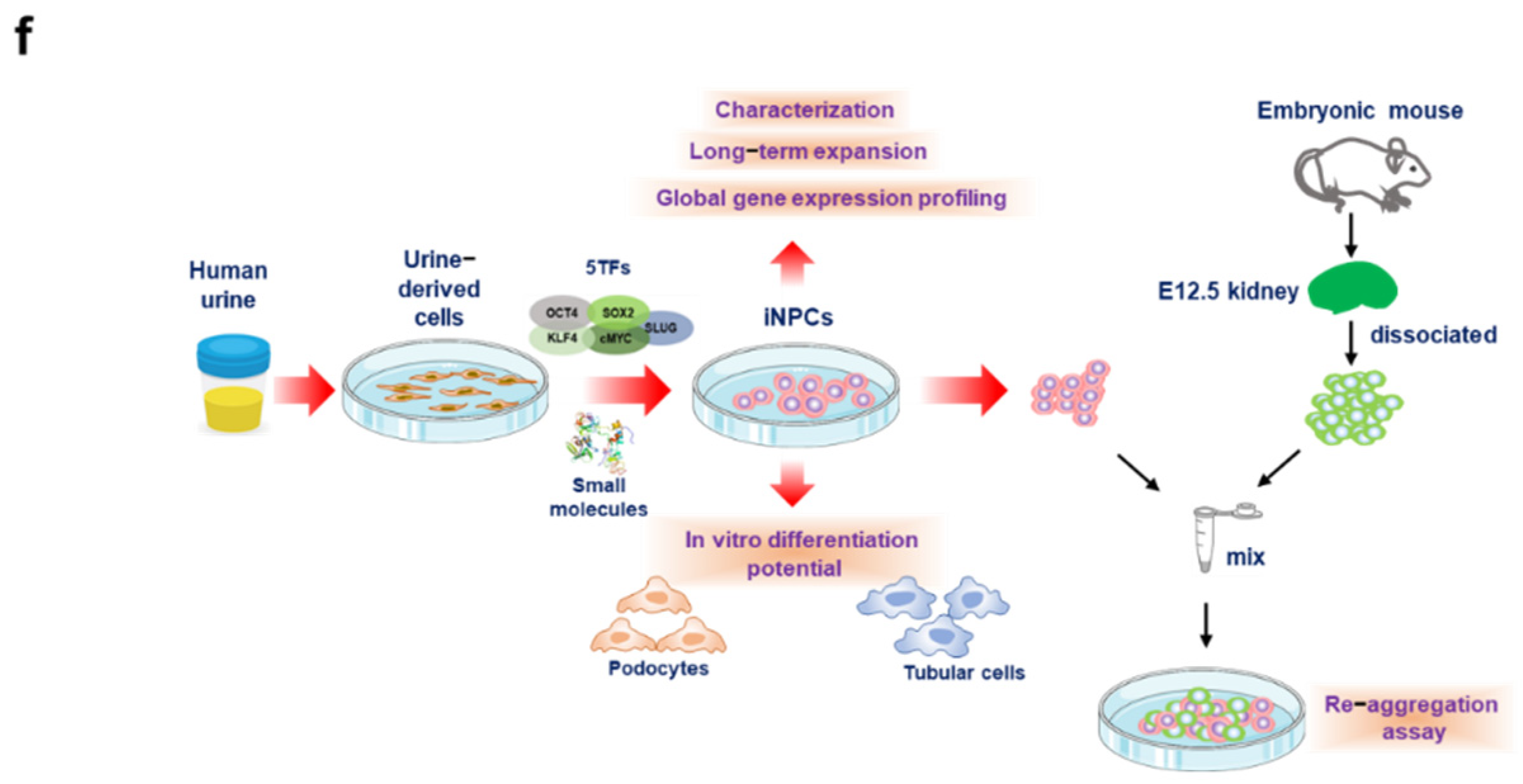
2.2. Generation of iNPCs from Human UCs
2.3. Characterization and In Vitro Expansion of iNPCs
2.4. Global Gene Expression Analysis of iNPCs
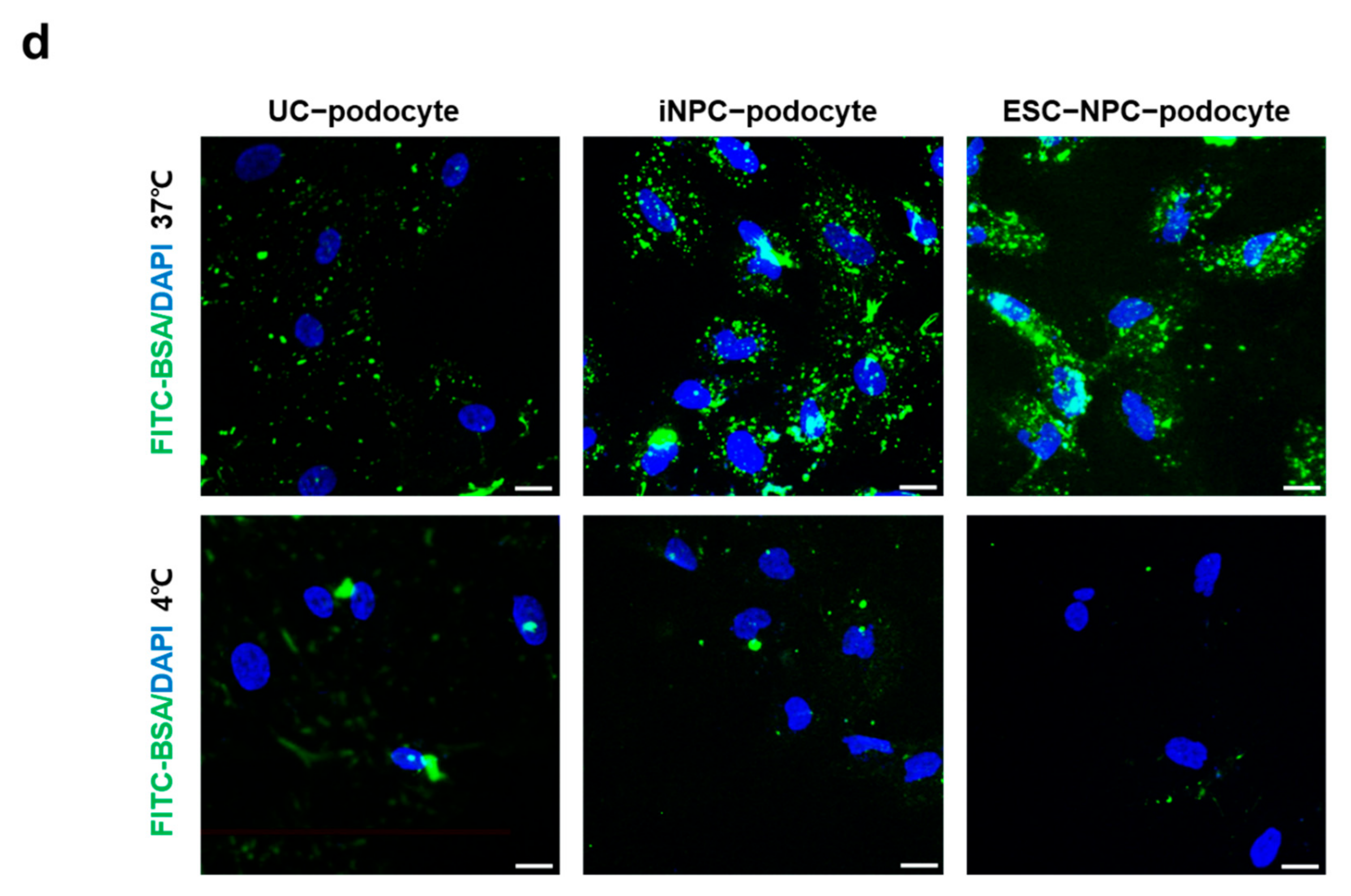
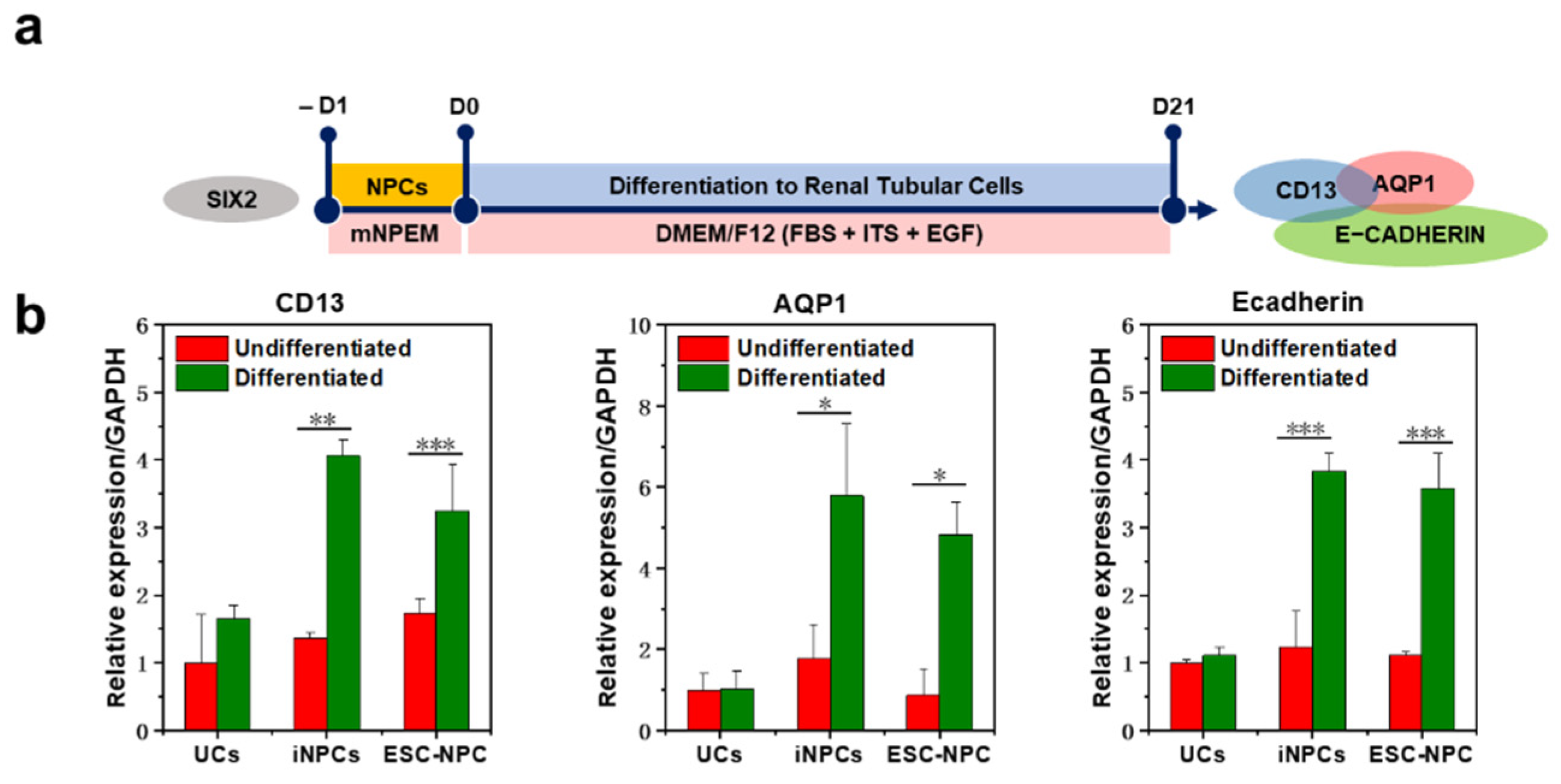
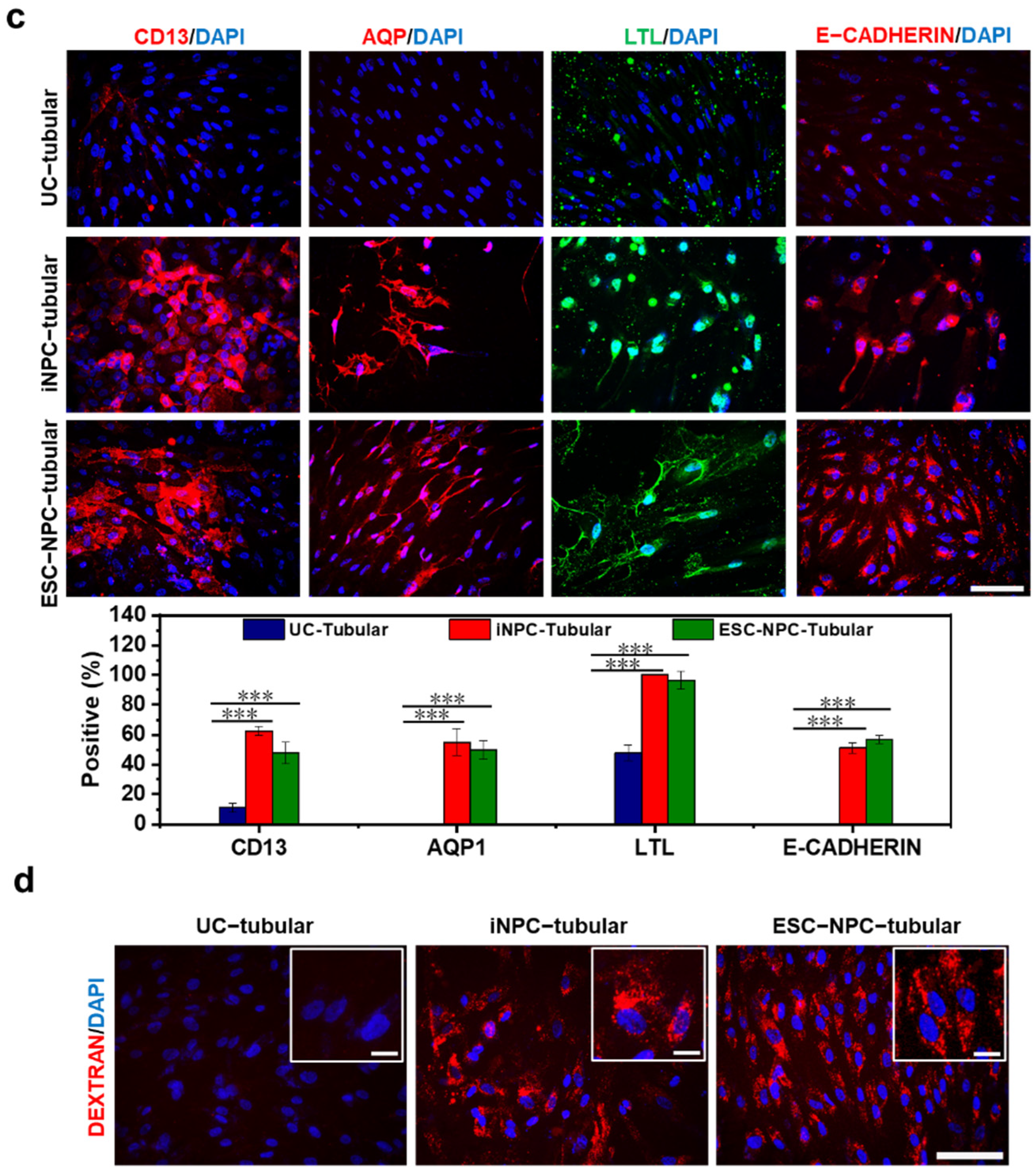
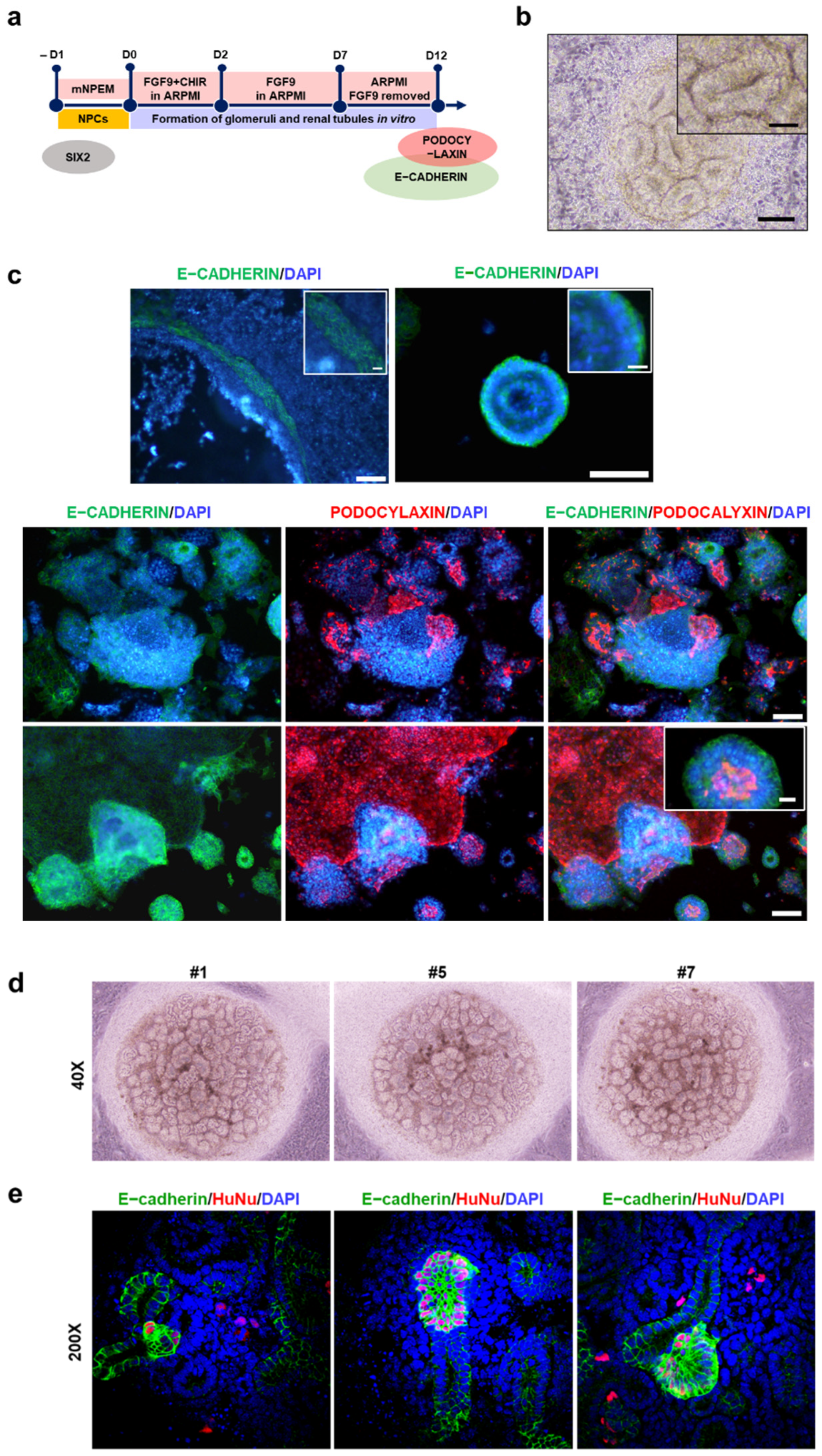
2.5. Differentiation Potential of iNPCs
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Human Urine Cells
4.2. Generation of iNPCs
4.3. In Vitro Differentiation Potential of iNPCs
4.4. Quantitative RT-PCR (qRT-PCR) and RT-PCR
4.5. Immunocytochemistry
4.6. Western Blot Analysis
4.7. RNA-Seq
4.8. Flow Cytometry
4.9. Karyotyping
4.10. Re-Aggregation Assay with Mouse Embryonic Kidney Cells
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bello, A.K.; Johnson, D.W.; Feehally, J.; Harris, D.; Jindal, K.; Lunney, M.; Okpechi, I.G.; Salako, B.L.; Wiebe, N.; Ye, F.; et al. Global Kidney Health Atlas (GKHA): Design and methods. Kidney Int. Suppl. 2017, 7, 145–153. [Google Scholar]
- De Nicola, L.; Zoccali, C. Chronic kidney disease prevalence in the general population: Heterogeneity and concerns. Nephrol. Dial. Transplant. 2016, 31, 331–335. [Google Scholar]
- Bantounas, I.; Ranjzad, P.; Tengku, F.; Silajdzic, E.; Forster, D.; Asselin, M.C.; Lewis, P.; Lennon, R.; Plagge, A.; Wang, Q.; et al. Generation of Functioning Nephrons by Implanting Human Pluripotent Stem Cell-Derived Kidney Progenitors. Stem Cell Rep. 2018, 10, 766–779. [Google Scholar]
- Brown, A.C.; Muthukrishnan, S.D.; Oxburgh, L. A synthetic niche for nephron progenitor cells. Dev. Cell 2015, 34, 229–241. [Google Scholar]
- Tanigawa, S.; Taguchi, A.; Sharma, N.; Perantoni, A.O.; Nishinakamura, R. Selective In Vitro Propagation of Nephron Progenitors Derived from Embryos and Pluripotent Stem Cells. Cell Rep. 2016, 15, 801–813. [Google Scholar]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar]
- Tanabe, K.; Haag, D.; Wernig, M. Direct somatic lineage conversion. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2015, 370, 368. [Google Scholar]
- Wang, H.; Yang, Y.; Liu, J.; Qian, L. Direct cell reprogramming: Approaches, mechanisms and progress. Nat. Rev. Mol. Cell Biol. 2021, 22, 410–424. [Google Scholar]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar]
- Yang, N.; Zuchero, J.B.; Ahlenius, H.; Marro, S.; Ng, Y.H.; Vierbuchen, T.; Hawkins, J.S.; Geissler, R.; Barres, B.A.; Wernig, M. Generation of oligodendroglial cells by direct lineage conversion. Nat. Biotechnol. 2013, 31, 434–439. [Google Scholar]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar]
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011, 475, 386–389. [Google Scholar]
- Efe, J.A.; Hilcove, S.; Kim, J.; Zhou, H.; Ouyang, K.; Wang, G.; Chen, J.; Ding, S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 2011, 13, 215–222. [Google Scholar]
- Margariti, A.; Winkler, B.; Karamariti, E.; Zampetaki, A.; Tsai, T.N.; Baban, D.; Ragoussis, J.; Huang, Y.; Han, J.D.; Zeng, L.; et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc. Natl. Acad. Sci. USA 2012, 109, 13793–13798. [Google Scholar]
- Kim, J.; Efe, J.A.; Zhu, S.; Talantova, M.; Yuan, X.; Wang, S.; Lipton, S.A.; Zhang, K.; Ding, S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 7838–7843. [Google Scholar]
- Ma, T.; Xie, M.; Laurent, T.; Ding, S. Progress in the reprogramming of somatic cells. Circ. Res. 2013, 112, 562–574. [Google Scholar]
- Hendry, C.E.; Vanslambrouck, J.M.; Ineson, J.; Suhaimi, N.; Takasato, M.; Rae, F.; Little, M.H. Direct transcriptional reprogramming of adult cells to embryonic nephron progenitors. J. Am. Soc. Nephrol. 2013, 24, 1424–1434. [Google Scholar]
- Vanslambrouck, J.M.; Woodard, L.E.; Suhaimi, N.; Williams, F.M.; Howden, S.E.; Wilson, S.B.; Lonsdale, A.; Er, P.X.; Li, J.; Maksimovic, J.; et al. Direct reprogramming to human nephron progenitor-like cells using inducible piggyBac transposon expression of SNAI2-EYA1-SIX1. Kidney Int. 2019, 95, 1153–1166. [Google Scholar]
- Zhao, M.T.; Chen, H.; Liu, Q.; Shao, N.Y.; Sayed, N.; Wo, H.T.; Zhang, J.Z.; Ong, S.G.; Liu, C.; Kim, Y.; et al. Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci. USA 2017, 114, E11111–E11120. [Google Scholar]
- Brunskill, E.W.; Aronow, B.J.; Georgas, K.; Rumballe, B.; Valerius, M.T.; Aronow, J.; Kaimal, V.; Jegga, A.G.; Yu, J.; Grimmond, S.; et al. Atlas of gene expression in the developing kidney at microanatomic resolution. Dev. Cell 2008, 15, 781–791. [Google Scholar]
- Lam, A.Q.; Freedman, B.S.; Morizane, R.; Lerou, P.H.; Valerius, M.T.; Bonventre, J.V. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 2014, 25, 1211–1225. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar]
- Morizane, R.; Bonventre, J.V. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat. Protoc. 2017, 12, 195–207. [Google Scholar]
- Kopan, R.; Chen, S.; Little, M. Nephron progenitor cells: Shifting the balance of self-renewal and differentiation. Curr. Top. Dev. Biol. 2014, 107, 293–331. [Google Scholar]
- Brophy, P.D.; Ostrom, L.; Lang, K.M.; Dressler, G.R. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development 2001, 128, 4747–4756. [Google Scholar]
- Morizane, R.; Lam, A.Q. Directed Differentiation of Pluripotent Stem Cells into Kidney. Biomark. Insights 2015, 10 (Suppl. S1), 147–152. [Google Scholar]
- Boyle, S.; Misfeldt, A.; Chandler, K.J.; Deal, K.K.; Southard-Smith, E.M.; Mortlock, D.P.; Baldwin, H.S.; de Caestecker, M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 2008, 313, 234–245. [Google Scholar]
- Mugford, J.W.; Yu, J.; Kobayashi, A.; McMahon, A.P. High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev. Biol. 2009, 333, 312–323. [Google Scholar]
- Da Sacco, S.; Thornton, M.E.; Petrosyan, A.; Lavarreda-Pearce, M.; Sedrakyan, S.; Grubbs, B.H.; De Filippo, R.E.; Perin, L. Direct Isolation and Characterization of Human Nephron Progenitors. Stem Cells Transl. Med. 2017, 6, 419–433. [Google Scholar]
- Harari-Steinberg, O.; Metsuyanim, S.; Omer, D.; Gnatek, Y.; Gershon, R.; Pri-Chen, S.; Ozdemir, D.D.; Lerenthal, Y.; Noiman, T.; Ben-Hur, H.; et al. Identification of human nephron progenitors capable of generation of kidney structures and functional repair of chronic renal disease. EMBO Mol. Med. 2013, 5, 1556–1568. [Google Scholar]
- Buzhor, E.; Omer, D.; Harari-Steinberg, O.; Dotan, Z.; Vax, E.; Pri-Chen, S.; Metsuyanim, S.; Pleniceanu, O.; Goldstein, R.S.; Dekel, B. Reactivation of NCAM1 defines a subpopulation of human adult kidney epithelial cells with clonogenic and stem/progenitor properties. Am. J. Pathol. 2013, 183, 1621–1633. [Google Scholar]
- Costantini, F.; Shakya, R. GDNF/Ret signaling and the development of the kidney. BioEssays News Rev. Mol. Cell. Dev. Biol. 2006, 28, 117–127. [Google Scholar]
- Ronconi, E.; Sagrinati, C.; Angelotti, M.L.; Lazzeri, E.; Mazzinghi, B.; Ballerini, L.; Parente, E.; Becherucci, F.; Gacci, M.; Carini, M.; et al. Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol. 2009, 20, 322–332. [Google Scholar]
- Van der Hauwaert, C.; Savary, G.; Gnemmi, V.; Glowacki, F.; Pottier, N.; Bouillez, A.; Maboudou, P.; Zini, L.; Leroy, X.; Cauffiez, C.; et al. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS ONE 2013, 8, e66750. [Google Scholar]
- Ciampi, O.; Iacone, R.; Longaretti, L.; Benedetti, V.; Graf, M.; Magnone, M.C.; Patsch, C.; Xinaris, C.; Remuzzi, G.; Benigni, A.; et al. Generation of functional podocytes from human induced pluripotent stem cells. Stem Cell Res. 2016, 17, 130–139. [Google Scholar]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar]
- Rauch, C.; Feifel, E.; Kern, G.; Murphy, C.; Meier, F.; Parson, W.; Beilmann, M.; Jennings, P.; Gstraunthaler, G.; Wilmes, A. Differentiation of human iPSCs into functional podocytes. PLoS ONE 2018, 13, e020386939. [Google Scholar]
- Qian, T.; Hernday, S.E.; Bao, X.; Olson, W.R.; Panzer, S.E.; Shusta, E.V.; Palecek, S.P. Directed Differentiation of Human Pluripotent Stem Cells to Podocytes under Defined Conditions. Sci. Rep. 2019, 9, 2765. [Google Scholar]
- Bajaj, P.; Rodrigues, A.D.; Steppan, C.M.; Engle, S.J.; Mathialagan, S.; Schroeter, T. Human Pluripotent Stem Cell-Derived Kidney Model for Nephrotoxicity Studies. Drug Metab. Dispos. 2018, 46, 1703–1711. [Google Scholar]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar]
- Prozialeck, W.C.; Lamar, P.C.; Appelt, D.M. Differential expression of E-cadherin, N-cadherin and beta-catenin in proximal and distal segments of the rat nephron. BMC Physiol. 2004, 4, 1–14. [Google Scholar]
- Unbekandt, M.; Davies, J.A. Dissociation of embryonic kidneys followed by reaggregation allows the formation of renal tissues. Kidney Int. 2010, 77, 407–416. [Google Scholar]
- Ganeva, V.; Unbekandt, M.; Davies, J.A. An improved kidney dissociation and reaggregation culture system results in nephrons arranged organotypically around a single collecting duct system. Organogenesis 2011, 7, 83–87. [Google Scholar]
- Xia, Y.; Sancho-Martinez, I.; Nivet, E.; Rodriguez Esteban, C.; Campistol, J.M.; Izpisua Belmonte, J.C. The generation of kidney organoids by differentiation of human pluripotent cells to ureteric bud progenitor-like cells. Nat. Protoc. 2014, 9, 2693–2704. [Google Scholar]
- Hartman, H.A.; Lai, H.L.; Patterson, L.T. Cessation of Renal Morphogenesis in Mice. Developmental biology 2007, 310, 379–387. [Google Scholar]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar]
- Berger, K.; Bangen, J.M.; Hammerich, L.; Liedtke, C.; Floege, J.; Smeets, B.; Moeller, M.J. Origin of regenerating tubular cells after acute kidney injury. Proc. Natl. Acad. Sci. USA 2014, 111, 1533–1538. [Google Scholar]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar]
- Liu, X.; Sun, H.; Qi, J.; Wang, L.; He, S.; Liu, J.; Feng, C.; Chen, C.; Li, W.; Guo, Y.; et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat. Cell Biol. 2013, 15, 829–838. [Google Scholar]
- Davies, J.A. Mesenchyme to epithelium transition during development of the mammalian kidney tubule. Acta Anat. 1996, 156, 187–201. [Google Scholar]
- Savagner, P.; Karavanova, I.; Perantoni, A.; Thiery, J.P.; Yamada, K.M. Slug mRNA Is Expressed by Specific Mesodermal Derivatives During Rodent Organogenesis. Dev. Dyn. 1998, 213, 182–187. [Google Scholar]
- Chang-Panesso, M.; Humphreys, B.D. Cellular plasticity in kidney injury and repair. Nature reviews. Nephrology 2017, 13, 39–46. [Google Scholar]
- Xu, J.; Du, Y.; Deng, H. Direct lineage reprogramming: Strategies, mechanisms, and applications. Cell Stem Cell 2015, 16, 119–134. [Google Scholar]
- Leuning, D.G.; Reinders, M.E.; Li, J.; Peired, A.J.; Lievers, E.; de Boer, H.C.; Fibbe, W.E.; Romagnani, P.; van Kooten, C.; Little, M.H.; et al. Clinical-Grade Isolated Human Kidney Perivascular Stromal Cells as an Organotypic Cell Source for Kidney Regenerative Medicine. Stem Cells Transl. Med. 2017, 6, 405–418. [Google Scholar]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar]
- Zheng, J.; Yun, W.; Park, J.; Kang, P.J.; Lee, G.; Song, G.; Kim, I.Y.; You, S. Long-term expansion of directly reprogrammed keratinocyte-like cells and in vitro reconstitution of human skin. J. Biomed. Sci. 2020, 27, 56. [Google Scholar]
- Kang, P.J.; Son, D.; Ko, T.H.; Hong, W.; Yun, W.; Jang, J.; Choi, J.I.; Song, G.; Lee, J.; Kim, I.Y.; et al. mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells. Cells 2019, 8, 1043. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, W.-W.; Zheng, J.; Yun, W.; Kang, P.-J.; Park, G.; Song, G.; Kim, I.-Y.; You, S. Generation of Induced Nephron Progenitor-like Cells from Human Urine-Derived Cells. Int. J. Mol. Sci. 2021, 22, 13449. https://doi.org/10.3390/ijms222413449
Gao W-W, Zheng J, Yun W, Kang P-J, Park G, Song G, Kim I-Y, You S. Generation of Induced Nephron Progenitor-like Cells from Human Urine-Derived Cells. International Journal of Molecular Sciences. 2021; 22(24):13449. https://doi.org/10.3390/ijms222413449
Chicago/Turabian StyleGao, Wei-Wei, Jie Zheng, Wonjin Yun, Phil-Jun Kang, Gyuman Park, Gwonhwa Song, In-Yong Kim, and Seungkwon You. 2021. "Generation of Induced Nephron Progenitor-like Cells from Human Urine-Derived Cells" International Journal of Molecular Sciences 22, no. 24: 13449. https://doi.org/10.3390/ijms222413449
APA StyleGao, W.-W., Zheng, J., Yun, W., Kang, P.-J., Park, G., Song, G., Kim, I.-Y., & You, S. (2021). Generation of Induced Nephron Progenitor-like Cells from Human Urine-Derived Cells. International Journal of Molecular Sciences, 22(24), 13449. https://doi.org/10.3390/ijms222413449