
Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation



Abstract
1. Introduction
2. Results
2.1. Mitochondrial Morphology Is Disrupted in a Neuronal Model of Polyglutamine Toxicity
2.2. Differences in Mitochondrial Morphology in Neuronal Model of Polyglutamine Toxicity Are Exacerbated with Increasing Age
2.3. Neuronal Model of Polyglutamine Toxicity Exhibits Altered Mitochondrial Function
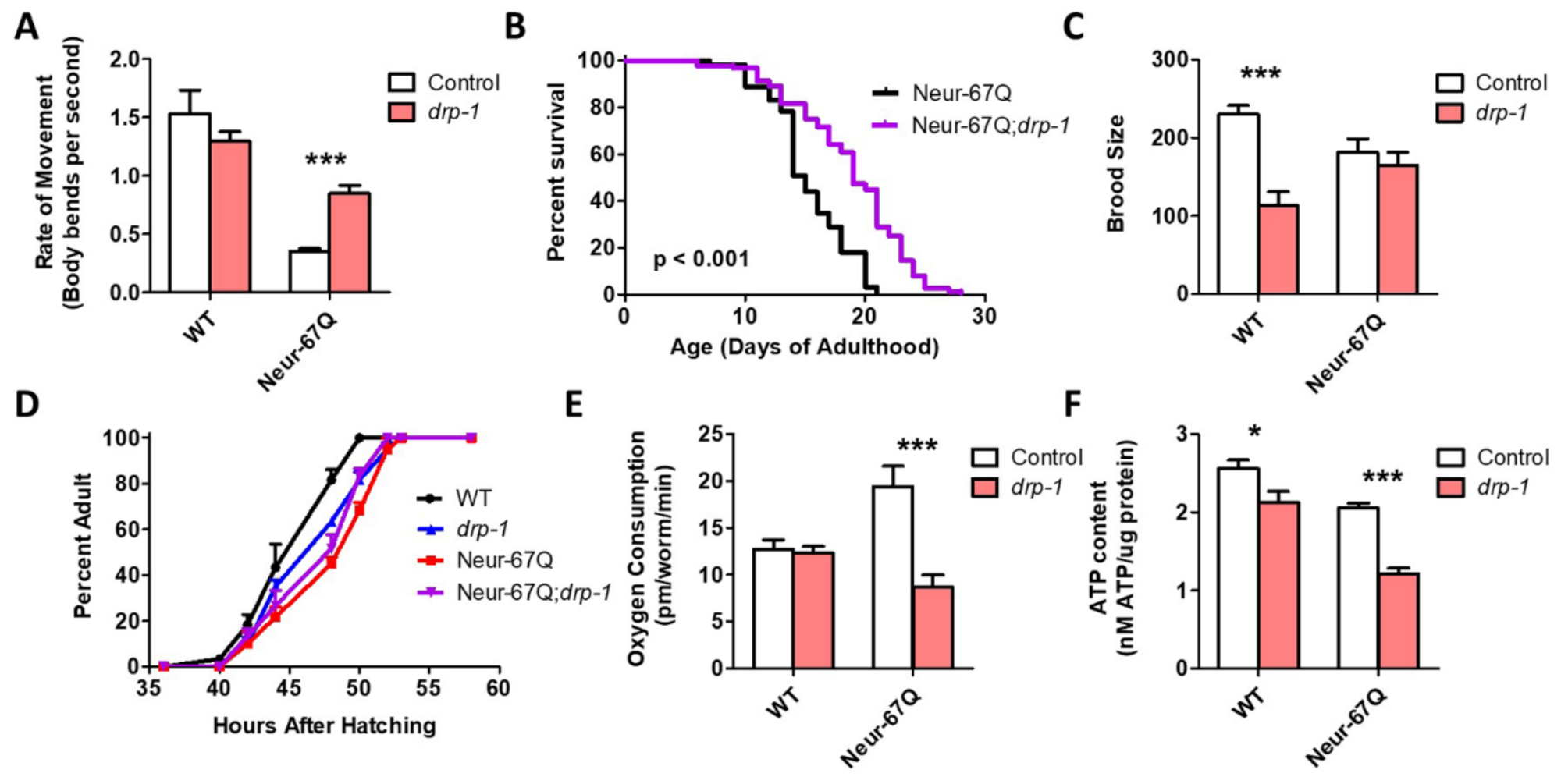
2.4. Disruption of Mitochondrial Fission Is Beneficial in a Neuronal Model of Polyglutamine Toxicity
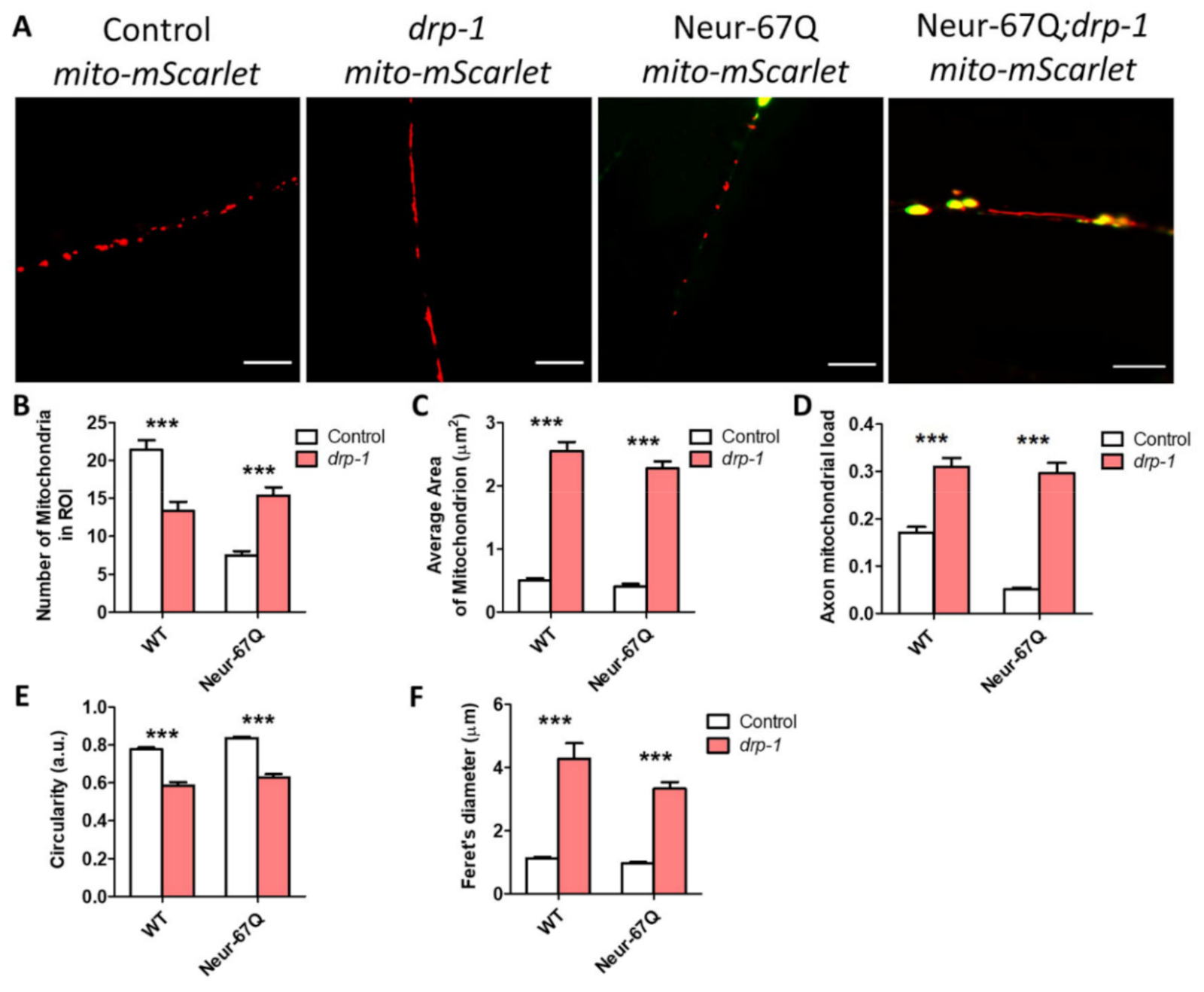
2.5. Disruption of Mitochondrial Fission Decreases Mitochondrial Fragmentation in Neurons
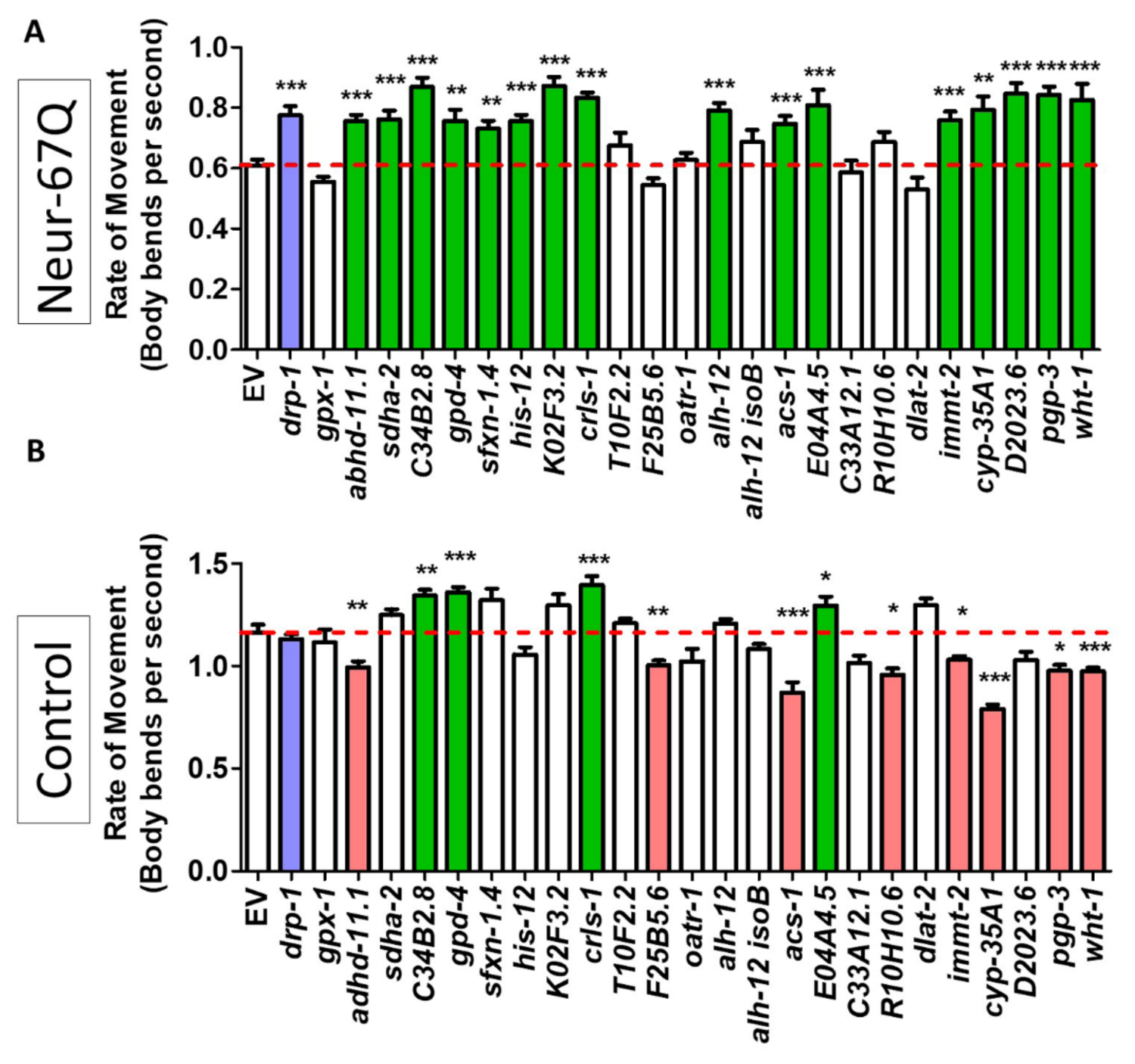
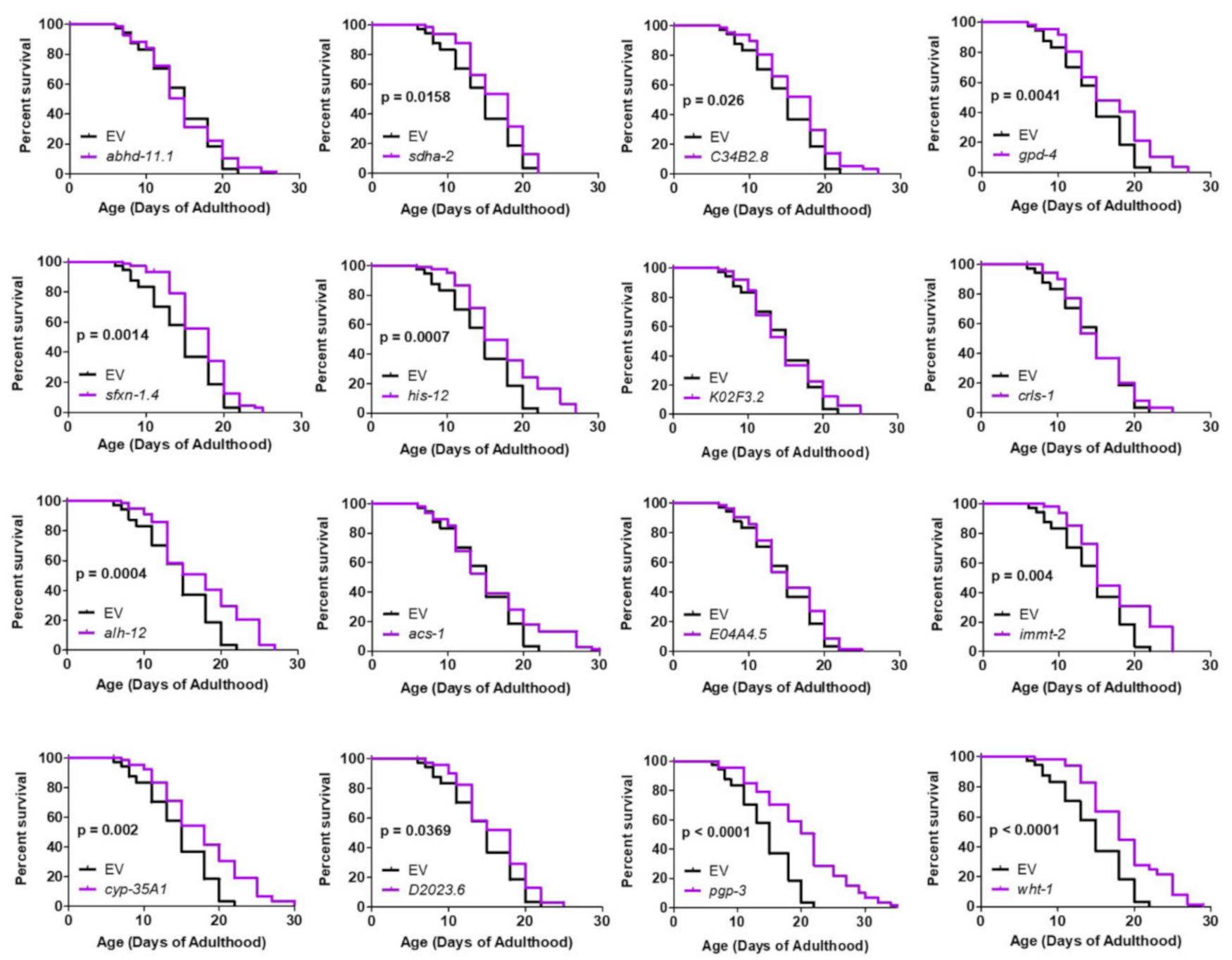
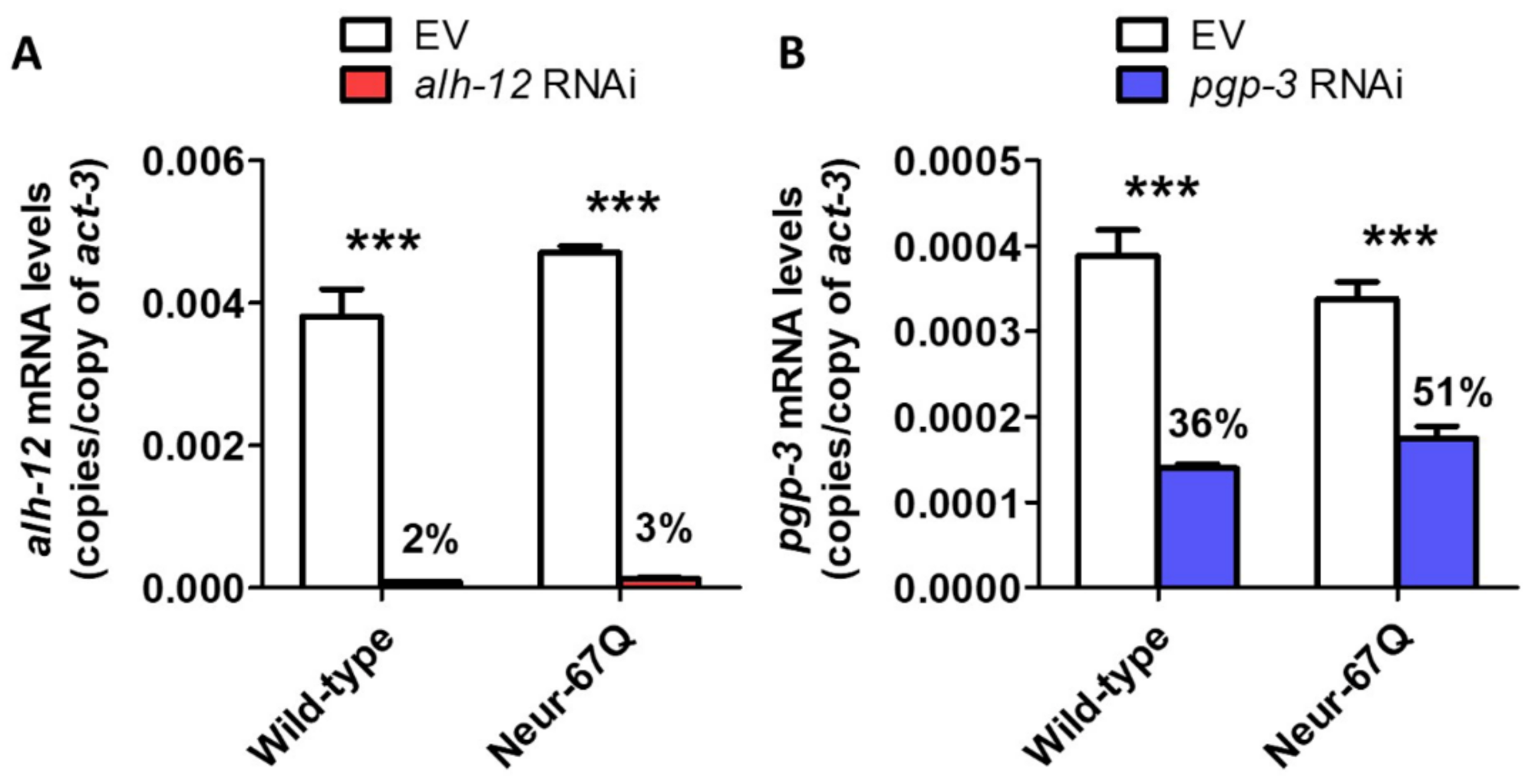
2.6. Targeting Genes That Affect Mitochondrial Fragmentation Improves Thrashing Rate and Lifespan in a Neuronal Model of Polyglutamine Toxicity
3. Discussion
3.1. CAG Repeat Expansion Disrupts Mitochondrial Morphology and Function in Neurons
3.2. Tissue-Specific Effects of Disrupting Mitochondrial Fission
3.3. Decreasing Mitochondrial Fragmentation as a Therapeutic Strategy for Polyglutamine Diseases
4. Materials and Methods
4.1. Strains
- N2 (WT)
- AM102 rmIs111[rgef-1p::40Q:YFP] referred to as Neur-40Q
- AM717 rmIs284[rgef-1p::67Q:YFP] referred to as Neur-67Q
- JVR258 drp-1(tm1108);rmIs284[rgef-1p::67Q:YFP]
- JVRV438 rmIs284[rgef-1p::67Q:YFP]; sid-1(pk3321); uIs69 [pCFJ90 (myo-2p::mCherry) + unc-119p::sid-1]
- JVR443 rmIs284[rgef-1p::67Q:YFP]; uIs69 [pCFJ90 (myo-2p::mCherry) + unc-119p::sid-1]
- PHX3820 sybIs3820[rab-3p::tomm-20::mScarlet] referred to as mito-mScarlet
- JVR611 rmIs284[rgef-1p::67Q:YFP];drp-1(tm1108); sybIs3820[rab-3p::tomm-20::mScarlet] referred to as Neur-67Q;drp-1;mito-mScarlet
- JVR612 rmIs284[rgef-1p::67Q:YFP]; sybIs3820[rab-3p::tomm-20::mScarlet] referred to as Neur-67Q;mito-mScarlet
- JVRV613 drp-1(tm1108); sybIs3820[rab-3p::tomm-20::mScarlet] referred to as drp-1;mito-mScarlet
- MQ17V53 drp-1 (tm1108)
- TU3401 sid-1(pk3321); uIs69 [pCFJ90 (myo-2p::mCherry) + unc-119p::sid-1]
4.2. Generation of Strains to Monitor Mitochondrial Morphology in GABA Neurons
4.3. Confocal Imaging and Quantification
4.4. Oxygen Consumption
4.5. ATP Production
4.6. Rate of Movement
4.7. Lifespan
4.8. Brood Size
4.9. Post-Embryonic Development
4.10. Quantitative Reverse-Transcription PCR (qPCR)
- yfp (L-GACGACGGCAACTACAAGAC, R-TCCTTGAAGTCGATGCCCTT);
- pgp-3 (L-CTGTCTGGTGGACAGAAGCA, R-AAGAGCTGACGTGGCTTCAT);
- alh-12 (L-GCCTTCAAGCTGGAACTGTTT, R-TTGCCTTTGTCTGAGTATGAGC).
4.11. RNAi
4.12. Experimental Design and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Lee, J.M.; Galkina, E.I.; Levantovsky, R.M.; Fossale, E.; Anne Anderson, M.; Gillis, T.; Srinidhi Mysore, J.; Coser, K.R.; Shioda, T.; Zhang, B.; et al. Dominant effects of the huntington’s disease htt cag repeat length are captured in gene-expression data sets by a continuous analysis mathematical modeling strategy. Hum. Mol. Genet. 2013, 22, 3227–3238. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, J.M.; Ramos, E.M.; Lee, J.H.; Gillis, T.; Mysore, J.S.; Hayden, M.R.; Warby, S.C.; Morrison, P.; Nance, M.; Ross, C.A.; et al. Cag repeat expansion in huntington disease determines age at onset in a fully dominant fashion. Neurology 2012, 78, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J. Nature and cause of mitochondrial dysfunction in Huntington’s disease: Focusing on huntingtin and the striatum. J. Neurochem. 2010, 114, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Wardman, J.H.; Hargreaves, I.P.; Neergheen, V.; Bie, A.S.; Tümer, Z.; Nielsen, J.E.; Nielsen, T.T. Evidence of oxidative stress and mitochondrial dysfunction in spinocerebellar ataxia type 2 (SCA2) patient fibroblasts: Effect of coenzyme Q10 supplementation on these parameters. Mitochondrion 2017, 34, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 27–42. [Google Scholar] [CrossRef]
- Lodi, R.; Schapira, A.H.; Manners, D.; Styles, P.; Wood, N.W.; Taylor, D.J.; Warner, T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000, 48, 72–76. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1h nmr spectroscopy. Neurology 1993, 43, 2689–2695. [Google Scholar] [CrossRef]
- Seong, I.S.; Ivanova, E.; Lee, J.M.; Choo, Y.S.; Fossale, E.; Anderson, M.; Gusella, J.F.; Laramie, J.M.; Myers, R.; Lesort, M.; et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005, 14, 2871–2880. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Trushina, E.; Dyer, R.B.; Badger, J.D., 2nd; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S.; et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Carbone, E.; Zhang, J.; Marzouk, E.; Villegas, M.; Siegel, A.; Nguyen, D.; Possidente, T.; Hartman, J.; Polley, K.; et al. Short-term succinic acid treatment mitigates cerebellar mitochondrial OXPHOS dysfunction, neurodegeneration and ataxia in a Purkinje-specific spinocerebellar ataxia type 1 (SCA1) mouse model. PLoS ONE 2017, 12, e0188425. [Google Scholar] [CrossRef]
- Kazachkova, N.; Raposo, M.; Montiel, R.; Cymbron, T.; Bettencourt, C.; Silva-Fernandes, A.; Duarte-Silva, S.; Maciel, P.; Lima, M. Patterns of mitochondrial DNA damage in blood and brain tissues of a transgenic mouse model of Machado-Joseph disease. Neurodegener. Dis. 2013, 11, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, E.; Yu, Z.; Chua, J.P.; Shimamura, R.; Zhao, L.; Zhu, F.; Venneti, S.; Pennuto, M.; Guan, Y.; Hung, G.; et al. Rescue of metabolic alterations in AR113Q skeletal muscle by peripheral androgen receptor gene silencing. Cell Rep. 2016, 17, 125–136. [Google Scholar] [CrossRef]
- Rocchi, A.; Milioto, C.; Parodi, S.; Armirotti, A.; Borgia, D.; Pellegrini, M.; Urciuolo, A.; Molon, S.; Morbidoni, V.; Marabita, M.; et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016, 132, 127–144. [Google Scholar] [CrossRef]
- Wang, H.; Lim, P.J.; Karbowski, M.; Monteiro, M.J. Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009, 18, 737–752. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef]
- Haun, F.; Nakamura, T.; Shiu, A.D.; Cho, D.H.; Tsunemi, T.; Holland, E.A.; La Spada, A.R.; Lipton, S.A. S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington’s disease. Antioxid. Redox Signal. 2013, 19, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease–associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Yu, Y.V.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant Huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef]
- Joshi, A.U.; Ebert, A.E.; Haileselassie, B.; Mochly-Rosen, D. Drp1/fis1-mediated mitochondrial fragmentation leads to lyso-somal dysfunction in cardiac models of Huntington’s disease. J. Mol. Cell. Cardiol. 2019, 127, 125–133. [Google Scholar] [CrossRef]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef]
- Machiela, E.; Rudich, P.D.; Traa, A.; Anglas, U.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Targeting mitochondrial network disorganization is protective in C. elegans models of Huntington’s disease. Aging Dis. 2021, 12, 1753–1772. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef] [PubMed]
- Borgia, D.; Malena, A.; Spinazzi, M.; Desbats, M.A.; Salviati, L.; Russell, A.P.; Miotto, G.; Tosatto, L.; Pegoraro, E.; Sorarù, G.; et al. Increased mitophagy in the skeletal muscle of spinal and bulbar muscular atrophy patients. Hum. Mol. Genet. 2017, 26, 1087–1103. [Google Scholar] [CrossRef]
- Harmuth, T.; Prell-Schicker, C.; Weber, J.J.; Gellerich, F.; Funke, C.; Driessen, S.; Magg, J.C.D.; Krebiehl, G.; Wolburg, H.; Hayer, S.N.; et al. Mitochondrial morphology, function and homeostasis are impaired by expression of an N-terminal calpain cleavage fragment of ataxin-3. Front. Mol. Neurosci. 2018, 11, 368. [Google Scholar] [CrossRef]
- Ward, J.M.; Stoyas, C.A.; Switonski, P.M.; Ichou, F.; Fan, W.; Collins, B.; Wall, C.E.; Adanyeguh, I.; Niu, C.; Sopher, B.L.; et al. Metabolic and organelle morphology defects in mice and human patients define spinocerebellar ataxia type 7 as a mitochondrial disease. Cell Rep. 2019, 26, 1189–1202. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Qi, X. Inhibition of drp1 hyperactivation reduces neuropathology and behavioral deficits in zq175 knock-in mouse model of huntington’s disease. Biochem. Biophys. Res. Commun. 2018, 507, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Sun, X.; Magpusao, A.; Fedorov, Y.; Thompson, M.; Wang, B.; Lundberg, K.; Adams, D.J.; Qi, X. Small-molecule sup-pression of calpastatin degradation reduces neuropathology in models of huntington’s disease. Nat. Commun. 2021, 12, 5305. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Carmo, C.; Campesan, S.; Fao, L.; Cotton, V.E.; Valero, J.; Lopes, C.; Rosenstock, T.R.; Giorgini, F.; Rego, A.C. Mi-tochondrial sirt3 confers neuroprotection in Huntington’s disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic. Biol. Med. 2021, 163, 163–179. [Google Scholar] [CrossRef]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.J.; Soh, M.S.; Chandhok, G.; Vijayaraghavan, T.; Teoh, J.S.; Crawford, S.; Cobham, A.E.; Yapa, N.M.B.; Mirth, C.K.; Neumann, B. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell. Mol. Life Sci. 2019, 76, 1967–1985. [Google Scholar] [CrossRef]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in dnm1l impairs mi-tochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Liontis, T.; Dues, D.J.; Rudich, P.D.; Traa, A.; Wyman, L.; Kaufman, C.; Cooper, J.F.; Lew, L.; Nadarajan, S.; et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. 2020, 34, 8475–8492. [Google Scholar] [CrossRef]
- Brignull, H.R.; Moore, F.E.; Tang, S.J.; Morimoto, R.I. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal caenorhabditis elegans model. J. Neurosci. 2006, 26, 7597–7606. [Google Scholar] [CrossRef]
- Firnhaber, C.; Hammarlund, M. Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. PLoS Genet. 2013, 9, e1003921. [Google Scholar] [CrossRef]
- Calixto, A.; Chelur, D.; Topalidou, I.; Chen, X.; Chalfie, M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 2010, 7, 554–559. [Google Scholar] [CrossRef]
- Ichishita, R.; Tanaka, K.; Sugiura, Y.; Sayano, T.; Mihara, K.; Oka, T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J. Biochem. 2008, 143, 449–454. [Google Scholar] [CrossRef]
- Huntington’s Study Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The huntington’s disease collaborative research group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Banfi, S.; Servadio, A.; Chung, M.Y.; Kwiatkowski, T.J.; McCall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.-Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lim, P.J.; Yin, C.; Rieckher, M.; Vogel, B.E.; Monteiro, M.J. Suppression of polyglutamine-induced toxicity in cell and animal models of huntington’s disease by ubiquilin. Hum. Mol. Genet. 2006, 15, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Connolly, J.B.; Wellington, C.; Hayden, M.; Dausset, J.; Neri, C. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc. Natl. Acad. Sci. USA 2001, 98, 13318–13323. [Google Scholar] [CrossRef]
- Nollen, E.A.; Garcia, S.M.; van Haaften, G.; Kim, S.; Chavez, A.; Morimoto, R.I.; Plasterk, R.H. From the cover: Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. USA 2004, 101, 6403–6408. [Google Scholar] [CrossRef]
- Lejeune, F.X.; Mesrob, L.; Parmentier, F.; Bicep, C.; Vazquez-Manrique, R.P.; Parker, J.A.; Vert, J.P.; Tourette, C.; Neri, C. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genom. 2012, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Salin, K.; Auer, S.K.; Rey, B.; Selman, C.; Metcalfe, N. Variation in the link between oxygen consumption and ATP production, and its relevance for animal performance. Proc. Biol. Sci. 2015, 282, 20151028. [Google Scholar] [CrossRef] [PubMed]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta BBA Bioenerg. 2010, 1797, 961–967. [Google Scholar] [CrossRef]
- Lemire, B.D.; Behrendt, M.; DeCorby, A.; Gaskova, D. C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech. Ageing Dev. 2009, 130, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.W.; Korasick, D.A.; Qureshi, I.A.; Campbell, A.C.; Gates, K.S.; Tanner, J.J. Inhibition, crystal structures, and in-solution oligomeric structure of aldehyde dehydrogenase 9A1. Biochem. Biophys. 2020, 691, 108477. [Google Scholar] [CrossRef]
- Lincke, C.R.; Broeks, A.; The, I.; Plasterk, R.H.; Borst, P. The expression of two P-glycoprotein (pgp) genes in transgenic Caenorhabditis elegans is confined to intestinal cells. EMBO J. 1993, 12, 1615–1620. [Google Scholar] [CrossRef]
- Mahajan-Miklos, S.; Tan, M.W.; Rahme, L.G.; Ausubel, F.M. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa–Caenorhabditis elegans pathogenesis model. Cell 1999, 96, 47–56. [Google Scholar] [CrossRef]
- Broeks, A.; Janssen, H.W.; Calafat, J.; Plasterk, R.H. A P-glycoprotein protects Caenorhabditis elegans against natural toxins. EMBO J. 1995, 14, 1858–1866. [Google Scholar] [CrossRef]
- Dues, D.J.; Andrews, E.K.; Schaar, C.E.; Bergsma, A.L.; Senchuk, M.M.; Van Raamsdonk, J.M. Aging causes decreased resistance to multiple stresses and a failure to activate specific stress response pathways. Aging 2016, 8, 777–795. [Google Scholar] [CrossRef]
- El Mouridi, S.; Lecroisey, C.; Tardy, P.; Mercier, M.; Leclercq-Blondel, A.; Zariohi, N.; Boulin, T. Reliable CRISPR/Cas9 Genome Engineering in Caenorhabditis elegans Using a Single Efficient sgRNA and an Easily Recognizable Phenotype. G3 Genes Genomes Genet. 2017, 7, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; Dejonge, H.; Blackwell, T.K.; et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Senchuk, M.M.; Van Raamsdonk, J.M. Delaying aging is neuroprotective in parkinson’s disease: A genetic analysis in C. elegans models. NPJ Parkinson’s Dis. 2015, 1, 15022. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. FUdR causes a twofold increase in the lifespan of the mitochondrial mutant gas-1. Mech. Ageing Dev. 2011, 132, 519–521. [Google Scholar] [CrossRef]
- Schaar, C.E.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Cooper, J.F.; Senchuk, M.; Hekimi, S.; Van Raamsdonk, J.M. Mitochondrial and cytoplasmic ros have opposing effects on lifespan. PLoS Genet. 2015, 11, e1004972. [Google Scholar] [CrossRef]
- Machiela, E.; Dues, D.J.; Senchuk, M.M.; Van Raamsdonk, J.M. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 2016, 96, 1–11. [Google Scholar] [CrossRef]
- Senchuk, M.M.; Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Madaj, Z.B.; Bowman, M.J.; Winn, M.E.; Van Raamsdonk, J.M. Activation of daf-16/foxo by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 2018, 14, e1007268. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Drosophila Homolog | Mammalian Homolog | Effect on Thrashing in Neur-67Q Worms | Effect on Lifespan in Neur-67Q Worms | Effect on Crawling in BW-Htt74Q Worms | Effect on Thrashing in BW-Htt74Q Worms | Effect on Thrashing in Neuron Specific RNAi Strain | Effect on Lifespan in Neuron Specific RNAi Strain | Effect of Crawling in BW-Htt28Q Worms | Effect of Thrashing in BW-Htt28Q Worms |
|---|---|---|---|---|---|---|---|---|---|---|
| alh-12 | Aldh | ALDH9A1 | Increased | Increased | Increased | Increased | No effect | Decreased | No effect | Decreased |
| pgp-3 | Mdr49 | ABCB4 | Increased | Increased | Increased | Increased | Decreased | Increased | No effect | Decreased |
| gpd-4 | Gapdh2 | GAPDH | Increased | Increased | Increased | No effect | Increased | No effect | No effect | Decreased |
| immt-2 | Mitofilin | IMMT | Increased | Increased | No effect | Increased | Decreased | Increased | No effect | No effect |
| sdha-2 | SdhA | SdhA | Increased | Increased | Increased | No effect | No effect | Increased | Increased | Decreased |
| wht-1 | w | ABCG1 | Increased | Increased | Increased | No effect | Decreased | Decreased | No effect | No effect |
| C34B2.8 | ND-B16.6 | NDUFA13 | Increased | Increased | Increased | Decreased | Increased | No effect | No effect | No effect |
| drp-1 | Drp1 | DNM1L | Increased | Increased | No effect | No effect | No effect | No effect | No effect | No effect |
| F25B5.6 | Fpgs | FPGS | No effect | ND | Increased | Increased | Decreased | ND | No effect | No effect |
| his-12 | His2A | HIS2H2AB | Increased | Increased | No effect | No effect | = | Decreased | Decreased | Decreased |
| sfxn-1.4 | Sfxn1-3 | SFXN1/3 | Increased | Increased | No effect | No effect | = | Decreased | No effect | No effect |
| abhd-11.1 | CG2059 | ABHD11 | Increased | = | No effect | No effect | Decreased | = | No effect | No effect |
| acs-1 | Acsf2 | ACSF2 | Increased | = | No effect | No effect | Decreased | = | Decreased | No effect |
| crls-1 | CLS | CRLS1 | Increased | = | No effect | No effect | Increased | = | No effect | No effect |
| cyp-35A1 | Cyp18a1 | CYP2C8 | Increased | Increased | Decreased | No effect | Decreased | = | No effect | No effect |
| D2023.6 | Adck1 | ADCK1 | Increased | Increased | Decreased | No effect | = | = | Decreased | No effect |
| dlat-2 | muc | DLAT | No effect | ND | Increased | No effect | No effect | ND | Decreased | Decreased |
| gpx-1 | PHGPx | GPX4 | No effect | ND | No effect | Increased | No effect | ND | No effect | No effect |
| timm-17B.1 | Tim17b | TIMM17A/B | Increased | = | No effect | No effect | Increased | = | No effect | Decreased |
| oatr-1 | Oat | OAT | No effect | ND | Increased | No effect | No effect | ND | Increased | No effect |
| R10H10.6 | CG2846 | RFK | No effect | ND | Increased | No effect | Decreased | ND | No effect | No effect |
| alh-12 iso B | Aldh | ALDH9A1 | = | ND | Decreased | No effect | = | ND | No effect | No effect |
| C33A12.1 | ND-13B | NDUFA5 | = | ND | No effect | No effect | = | ND | No effect | No effect |
| K02F3.2 | aralar1 | SLC25A12 | Increased | = | Decreased | No effect | = | = | No effect | Decreased |
| T10F2.2 | CG1628 | SLC25A15 | = | ND | No effect | No effect | = | ND | No effect | No effect |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traa, A.; Machiela, E.; Rudich, P.D.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation. Int. J. Mol. Sci. 2021, 22, 13447. https://doi.org/10.3390/ijms222413447
Traa A, Machiela E, Rudich PD, Soo SK, Senchuk MM, Van Raamsdonk JM. Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation. International Journal of Molecular Sciences. 2021; 22(24):13447. https://doi.org/10.3390/ijms222413447
Chicago/Turabian StyleTraa, Annika, Emily Machiela, Paige D. Rudich, Sonja K. Soo, Megan M. Senchuk, and Jeremy M. Van Raamsdonk. 2021. "Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation" International Journal of Molecular Sciences 22, no. 24: 13447. https://doi.org/10.3390/ijms222413447
APA StyleTraa, A., Machiela, E., Rudich, P. D., Soo, S. K., Senchuk, M. M., & Van Raamsdonk, J. M. (2021). Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation. International Journal of Molecular Sciences, 22(24), 13447. https://doi.org/10.3390/ijms222413447