
New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
2.1. Study Cohort
2.2. WES and Variants Pathogenicity Assessment
2.3. Clinical Features of Patients with Truncating Variants of ID/ASD Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Selection of Patients and DNA Samples Preparation
4.2. Whole-Exome Sequencing
4.3. Filtering and Variant Prioritization
4.4. Sanger Sequencing
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- May, M.E.; Kennedy, C.H. Health and Problem Behavior Among People with Intellectual Disabilities. Behav. Anal. Pract. 2010, 3, 4–12. [Google Scholar] [CrossRef]
- Bishop, S.L.; Farmer, C.; Thurm, A. Measurement of Nonverbal IQ in Autism Spectrum Disorder: Scores in Young Adulthood Compared to Early Childhood. J. Autism Dev. Disord. 2015, 45, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Kiani, A.K.; Miertus, J.; Barati, S.; Manara, E.; Paolacci, S.; Stuppia, L.; Gurrieri, F.; Bertelli, M. Genetic Analysis of Intellectual Disability and Autism. Acta Biomed. 2020, 9, 91. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schwartz, C.E. Intellectual Disability and Autism Spectrum Disorders: Causal Genes and Molecular Mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [PubMed]
- la Malfa, G.; Lassi, S.; Bertelli, M.; Salvini, R.; Placidi, G.F. Autism and Intellectual Disability: A Study of Prevalence on a Sample of the Italian Population. J. Intellect. Disabil. Res. 2004, 48, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Hodges, H.; Fealko, C.; Soares, N. Autism Spectrum Disorder: Definition, Epidemiology, Causes, and Clinical Evaluation. Transl. Pediatrics 2020, 9, S55. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism Spectrum Disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Moeschler, J.B.; Shevell, M.; Saul, R.A.; Chen, E.; Freedenberg, D.L.; Hamid, R.; Jones, M.C.; Stoler, J.M.; Tarini, B.A. Comprehensive Evaluation of the Child with Intellectual Disability or Global Developmental Delays. Pediatrics 2014, 134, e903–e918. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism Risk Factors: Genes, Environment, and Gene-Environment Interactions. Dialogues Clin. Neurosci. 2012, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the Genetic Architecture to Synaptic Plasticity in Autism Spectrum Disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Dulac, C. Brain Function and Chromatin Plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatrics 2021, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.L.; Vitobello, A.; Tran Mau-Them, F.; Nambot, S.; Sorlin, A.; Denommé-Pichon, A.S.; Delanne, J.; Moutton, S.; Callier, P.; Duffourd, Y.; et al. Next-Generation Sequencing Approaches and Challenges in the Diagnosis of Developmental Anomalies and Intellectual Disability. Clin. Genet. 2020, 98, 433–444. [Google Scholar] [CrossRef]
- Harripaul, R.; Noor, A.; Ayub, M.; Vincent, J.B. The Use of Next-Generation Sequencing for Research and Diagnostics for Intellectual Disability. Cold Spring Harb. Perspect. Med. 2017, 7, a026864. [Google Scholar] [CrossRef] [PubMed]
- Valentino, F.; Bruno, L.P.; Doddato, G.; Giliberti, A.; Tita, R.; Resciniti, S.; Fallerini, C.; Bruttini, M.; lo Rizzo, C.; Mencarelli, M.A.; et al. Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sci. 2021, 11, 936. [Google Scholar] [CrossRef]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; et al. Impact of On-Site Clinical Genetics Consultations on Diagnostic Rate in Children and Young Adults with Autism Spectrum Disorder. Mol. Autism 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ontario Health (Quality). Genome-Wide Sequencing for Unexplained Developmental Disabilities or Multiple Congenital Anomalies: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2020, 20, 1–178. [Google Scholar]
- Hewson, S.; Puka, K.; Mercimek-Mahmutoglu, S. Variable expressivity of a likely pathogenic variant in KCNQ2 in a three-generation pedigree presenting with intellectual disability with childhood onset seizures. Am. J. Med. Genet. A 2017, 173, 2226–2230. [Google Scholar] [CrossRef]
- Stessman, H.A.F.; Willemsen, M.H.; Fenckova, M.; Penn, O.; Hoischen, A.; Xiong, B.; Wang, T.; Hoekzema, K.; Vives, L.; Vogel, I.; et al. Disruption of POGZ Is Associated with Intellectual Disability and Autism Spectrum Disorders. Am. J. Hum. Genet. 2016, 98, 541–552. [Google Scholar] [CrossRef]
- Helbig, K.L.; Farwell Hagman, K.D.; Shinde, D.N.; Mroske, C.; Powis, Z.; Li, S.; Tang, S.; Helbig, I. Diagnostic Exome Sequencing Provides a Molecular Diagnosis for a Significant Proportion of Patients with Epilepsy. Genet. Med. 2016, 18, 898–905. [Google Scholar] [CrossRef]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci. Bull. 2017, 33, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Lenroot, R.K.; Yeung, P.K. Heterogeneity within Autism Spectrum Disorders: What Have We Learned from Neuroimaging Studies? Front. Hum. Neurosci. 2013, 7, 733. [Google Scholar] [CrossRef] [PubMed]
- Barbarese, E.; Barry, C.; Chou, C.J.; Goldstein, D.J.; Nakos, G.A.; Hyde-DeRuyscher, R.; Scheld, K.; Carson, J.H. Expression and Localization of Myelin Basic Protein in Oligodendrocytes and Transfected Fibroblasts. J. Neurochem. 1988, 51, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Thatikunta, P.; Steplewski, A.; Johnson, E.M.; Khalili, K.; Amini, S. A 39-KD DNA-Binding Protein from Mouse Brain Stimulates Transcription of Myelin Basic Protein Gene in Oligodendrocytic Cells. J. Cell Biol. 1995, 130, 1171–1179. [Google Scholar] [CrossRef]
- Roach, A.; Boylan, K.; Horvath, S.; Prusiner, S.B.; Hood, L.E. Characterization of Cloned CDNA Representing Rat Myelin Basic Protein: Absence of Expression in Brain of Shiverer Mutant Mice. Cell 1983, 34, 799–806. [Google Scholar] [CrossRef]
- Kamholz, J.; Toffenetti, J.; Lazzarini, R.A. Organization and Expression of the Human Myelin Basic Protein Gene. J. Neurosci. Res. 1988, 21, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Ribich, S.; Tasic, B.; Maniatis, T. Identification of Long-Range Regulatory Elements in the Protocadherin-α Gene Cluster. Proc. Natl. Acad. Sci. USA 2006, 103, 19719–19724. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Hamada, S.; Kumode, Y.; Esumi, S.; Katori, S.; Fukuda, E.; Uchiyama, Y.; Hirabayashi, T.; Mombaerts, P.; Yagi, T. The Protocadherin-α Family Is Involved in Axonal Coalescence of Olfactory Sensory Neurons into Glomeruli of the Olfactory Bulb in Mouse. Mol. Cell. Neurosci. 2008, 38, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, E.; Hamada, S.; Hasegawa, S.; Katori, S.; Sanbo, M.; Miyakawa, T.; Yamamoto, T.; Yamamoto, H.; Hirabayashi, T.; Yagi, T. Down-Regulation of Protocadherin-α A Isoforms in Mice Changes Contextual Fear Conditioning and Spatial Working Memory. Eur. J. Neurosci. 2008, 28, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Riazuddin, S.; Bernstein, S.L.; Ahmed, Z.; Khan, S.; Griffith, A.J.; Morell, R.J.; Friedman, T.B.; Riazuddin, S.; Wilcox, E.R. Mutations of the Protocadherin Gene PCDH15 Cause Usher Syndrome Type 1F. Am. J. Hum. Genet. 2001, 69, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Rebibo-Sabbah, A.; Nudelman, I.; Ahmed, Z.M.; Baasov, T.; Ben-Yosef, T. In Vitro and Ex Vivo Suppression by Aminoglycosides of PCDH15 Nonsense Mutations Underlying Type 1 Usher Syndrome. Hum. Genet. 2007, 122, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.M.; Riazuddin, S.; Aye, S.; Ali, R.A.; Venselaar, H.; Anwar, S.; Belyantseva, P.P.; Qasim, M.; Riazuddin, S.; Friedman, T.B. Gene Structure and Mutant Alleles of PCDH15: Nonsyndromic Deafness DFNB23 and Type 1 Usher Syndrome. Hum. Genet. 2008, 124, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Carvill, S. Sensory Impairments, Intellectual Disability and Psychiatry. J. Intellect. Disabil. Res. 2001, 45, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Domanico, D.; Fragiotta, S.; Trabucco, P.; Nebbioso, M.; Vingolo, E.M. Genetic Analysis for Two Italian Siblings with Usher Syndrome and Schizophrenia. Case Rep. Ophthalmol. Med. 2012, 2012, 380863. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.P.; DaNivas, V.; Venkatasubramanian, G.; Behere, R.V.; Gangadhar, B.N. Comorbid Bipolar Disorder and Usher Syndrome. Prim. Care Companion J. Clin. Psychiatry 2010, 12, PCC.09l00792. [Google Scholar] [CrossRef]
- Rijavec, N.; Grubic, V.N. Usher Syndrome and Psychiatric Symptoms: A Challenge in Psychiatric Management. Psychiatr. Danub. 2009, 21, 68–71. [Google Scholar]
- Ohara, O.; Nagase, T.; Mitsui, G.; Kohga, H.; Kikuno, R.; Hiraoka, S.; Takahashi, Y.; Kitajima, S.; Saga, Y.; Koseki, H. Characterization of Size-Fractionated CDNA Libraries Generated by the in Vitro Recombination-Assisted Method. DNA Res. 2002, 9, 47–57. [Google Scholar] [CrossRef][Green Version]
- Lawson, J.E.; Park, S.H.; Mattison, A.R.; Yan, J.; Reed, L.J. Cloning, Expression, and Properties of the Regulatory Subunit of Bovine Pyruvate Dehydrogenase Phosphatase. J. Biol. Chem. 1997, 272, 31625–31629. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; di Lullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De Novo Mutations Revealed by Whole-Exome Sequencing Are Strongly Associated with Autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, Z.; Liu, Z.; Zhang, N.; Wang, R.; Li, F.; Zhang, T.; Jiang, Y.; Zhi, X.; Wang, Z.; et al. Nonrandom Occurrence of Multiple de Novo Coding Variants in a Proband Indicates the Existence of an Oligogenic Model in Autism. Genet. Med. 2020, 22, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Wilfert, A.B.; Turner, T.N.; Murali, S.C.; Hsieh, P.H.; Sulovari, A.; Wang, T.; Coe, B.P.; Guo, H.; Hoekzema, K.; Bakken, T.E.; et al. Recent Ultra-Rare Inherited Variants Implicate New Autism Candidate Risk Genes. Nat. Genet. 2021, 53, 1125–1134. [Google Scholar] [CrossRef]
- Coll-Tane, M.; Krebbers, A.; Castells-Nobau, A.; Zweier, C.; Schenck, A. Intellectual Disability and Autism Spectrum Disorders “on the Fly”: Insights from Drosophila. DMM Dis. Models Mech. 2019, 12, dmm039180. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Venselaar, H.; te Beek, T.A.H.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein Structure Analysis of Mutations Causing Inheritable Diseases. An e-Science Approach with Life Scientist Friendly Interfaces. BMC Bioinform. 2010, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Proband | Gene | Transcript (hg19) | Variant (HGVS) | Protein (HGVS) | MAF (gnomAD All) | MAF (gnomAD NFE) | dbSNP | ClinVar Classification | CADD | Transmission | Origin | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | MBP | NM_001025081.1 | c.138del | p.(Phe46Leufs * 18) | NA | NA | NA | NA | NA | Autosomal dominant | De novo | Pathogenic |
| II | PCDH15 | NM_033056.3 | c.5573_5576dup | p.(Lys1859Asnfs * 2) | 0.0068% | 0.011% | rs770082088 | NA | NA | Autosomal dominant | De novo | Pathogenic |
| III | PCDHA1 | NM_018900.3 | c.670_673dup | p.(Thr225Argfs * 4) | NA | NA | NA | NA | NA | Autosomal dominant | De novo | Pathogenic |
| IV | PDPR | NM_001322118.1 | c.826C > T | p.(Gln276*) | 0.00071% | 0.0016% | NA | NA | 22.7 | Autosomal dominant | De novo | Pathogenic |
| Proband | Gene | Transcript (hg19) | Variant (HGVS) | Protein (HGVS) | MAF (gnomAD All) | MAF (gnomADNFE) | dbSNP | ClinVar Classification | CADD | Transmission | Origin | Classification | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VIII | ACTB | NM_001101.4 | c.583G > A | p.(Glu195Lys) | NA | NA | NA | Likely pathogenic | 37 | Autosomal dominant | De novo | Likely pathogenic | NA |
| IX | KCNQ2 | NM_172107.2 | c.628C > T | p.(Arg210Cys) | NA | NA | rs796052626 | Pathogenic | 27.3 | Autosomal dominant | De novo | Pathogenic | [20] |
| X | KMT2A | NM_001197104.1 | c.478C > T | p.(Arg160*) | NA | NA | NA | NA | 36 | Autosomal dominant | De novo | Pathogenic | NA |
| XI | PACS1 | NM_018026.3 | c.607C > T | p.(Arg203Trp) | 0% | NA | rs398123009 | Pathogenic | 29.4 | Autosomal dominant | De novo | Pathogenic | [21] |
| XII | POGZ | NM_145796.3 | c.2716C > T | p.(Arg906 *) | NA | NA | rs869312833 | Pathogenic | 12.48 | Autosomal dominant | De novo | Pathogenic | [22] |
| XIII | SHANK3 | NM_001080420.1 | c.1807_1811del | p.(Val604Leufs * 80) | NA | NA | NA | NA | NA | Autosomal dominant | De novo | Pathogenic | NA |
| XIV | SMAD6 | NM_005585.4 | c.137dup | p.(Tyr459Leufs * 106) | NA | NA | NA | NA | NA | Autosomal dominant | De novo | Pathogenic | NA |
| XV | SYNGAP1 | NM_001130066.1 | c.3670C > T | p.(Arg1224 *) | NA | NA | rs869312955 | Pathogenic | 36 | Autosomal dominant | De novo | Pathogenic | [23] |
| Proband | Gene | Variant (HGVS) | Protein (HGVS) | A Gender | Age (Years Old) | ID/ASD | Craniofacial Dysmorphisms | Additional Clinical Signs |
|---|---|---|---|---|---|---|---|---|
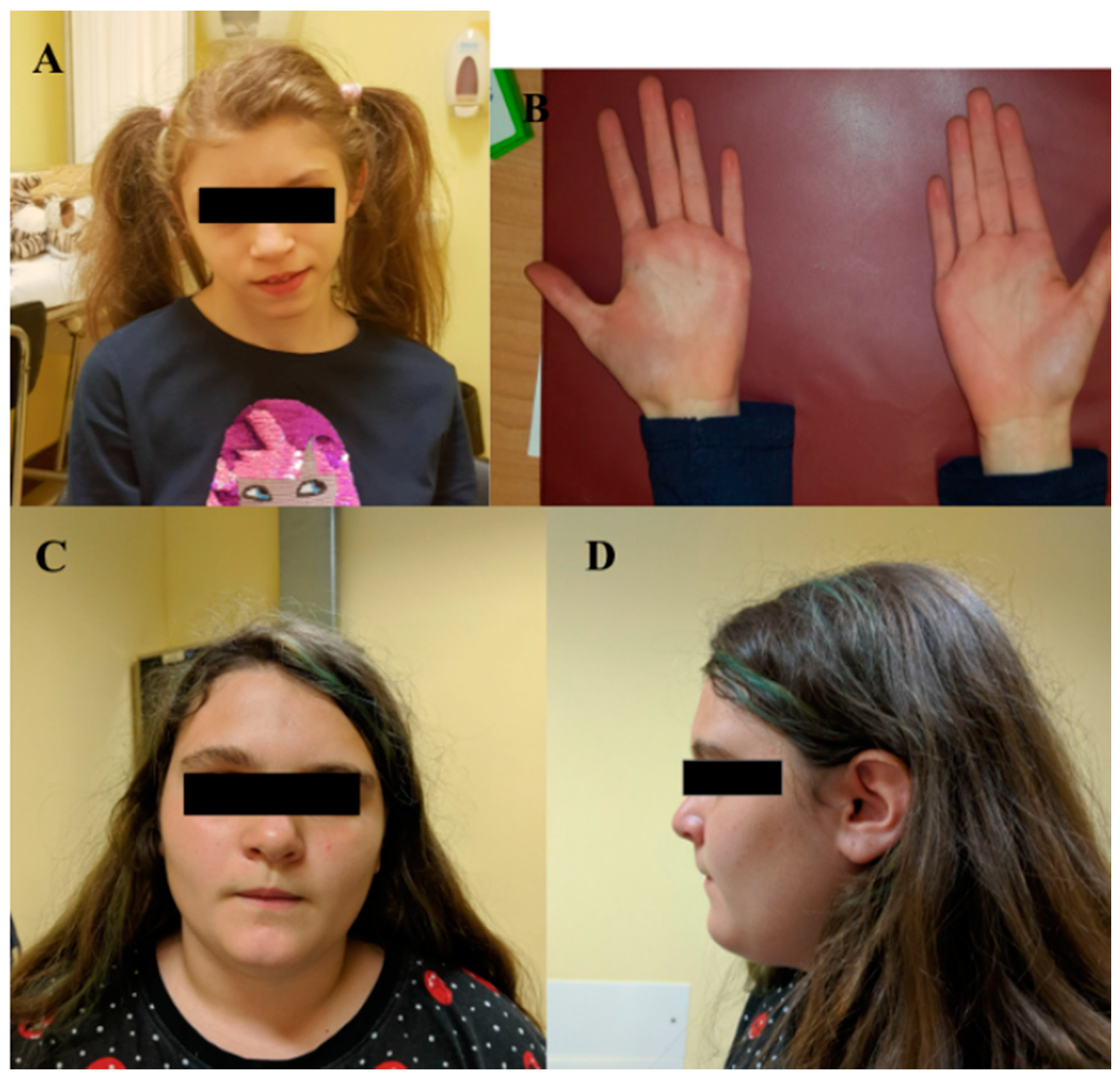
| I | MBP | c.138del | p.(Phe46Leufs*18) | F | 15 | ID | Triangular facies, prominent ears, thin upper lip, absent eyebrows, broad nasal bridge, bulbous nasal tip, thin and sparse hair, everted lower lip, M-shaped upper lip, hairline anteriorly advanced. | Hyperactivity, language delay and aggressiveness, disturbed wake–sleep cycle, arachnodactyly of the hand and feet. |
| II | PCDH15 | c.5573_5576dup | p.(Lys1859Asnfs * 2) | F | 14 | ID | Square-shaped face, deeply set eyes, bilateral underfolded helix, short and stocky neck. | Epilepsy, language and psychomotor delay. |
| III | PCDHA1 | c.670_673dup | p.(Thr225Argfs * 4) | M | 9 | ASD and ID | Simplified auricles | ADHD, vermis and brain stem hypoplasia, tortuous course of the optic nerves, flat feet, hyperlaxity, psychomotor and language delay. |
| IV | PDPR | c.826C > T | p.(Gln276*) | M | 11 | ID | Deep-set eyes, wide nasal tip, thin upper lip, chin dimple, and macrodontia. | Cognitive impairment, repetitive behaviors, an altered sleep pattern with difficulty in falling asleep, isolationist tendencies, manual stereotypies, hypochromic stains, food selectiveness, language and psychomotor delay. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, L.P.; Doddato, G.; Valentino, F.; Baldassarri, M.; Tita, R.; Fallerini, C.; Bruttini, M.; Lo Rizzo, C.; Mencarelli, M.A.; Mari, F.; et al. New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. Int. J. Mol. Sci. 2021, 22, 13439. https://doi.org/10.3390/ijms222413439
Bruno LP, Doddato G, Valentino F, Baldassarri M, Tita R, Fallerini C, Bruttini M, Lo Rizzo C, Mencarelli MA, Mari F, et al. New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. International Journal of Molecular Sciences. 2021; 22(24):13439. https://doi.org/10.3390/ijms222413439
Chicago/Turabian StyleBruno, Lucia Pia, Gabriella Doddato, Floriana Valentino, Margherita Baldassarri, Rossella Tita, Chiara Fallerini, Mirella Bruttini, Caterina Lo Rizzo, Maria Antonietta Mencarelli, Francesca Mari, and et al. 2021. "New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing" International Journal of Molecular Sciences 22, no. 24: 13439. https://doi.org/10.3390/ijms222413439
APA StyleBruno, L. P., Doddato, G., Valentino, F., Baldassarri, M., Tita, R., Fallerini, C., Bruttini, M., Lo Rizzo, C., Mencarelli, M. A., Mari, F., Pinto, A. M., Fava, F., Fabbiani, A., Lamacchia, V., Carrer, A., Caputo, V., Granata, S., Benetti, E., Zguro, K., ... Ariani, F. (2021). New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. International Journal of Molecular Sciences, 22(24), 13439. https://doi.org/10.3390/ijms222413439