
Electron Holes in G-Quadruplexes: The Role of Adenine Ending Groups


 and
and 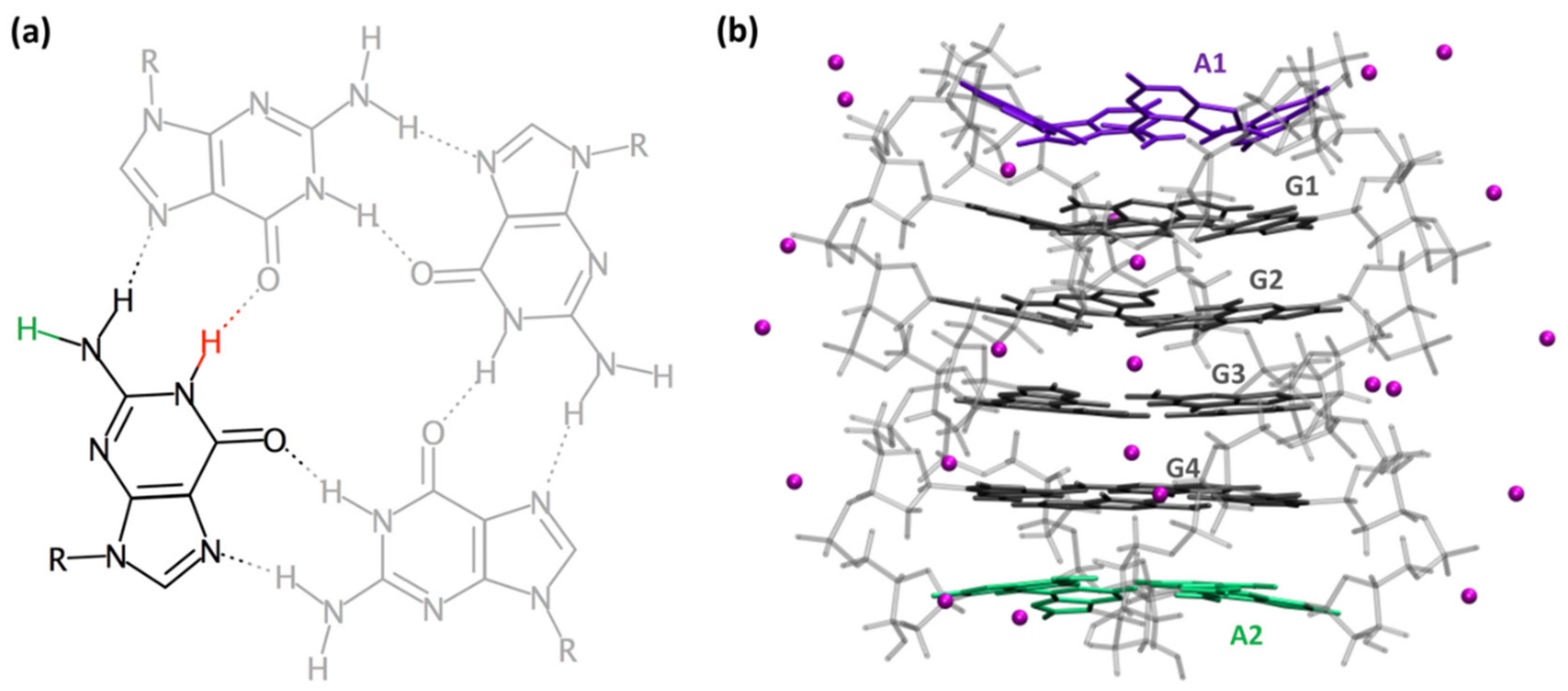{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
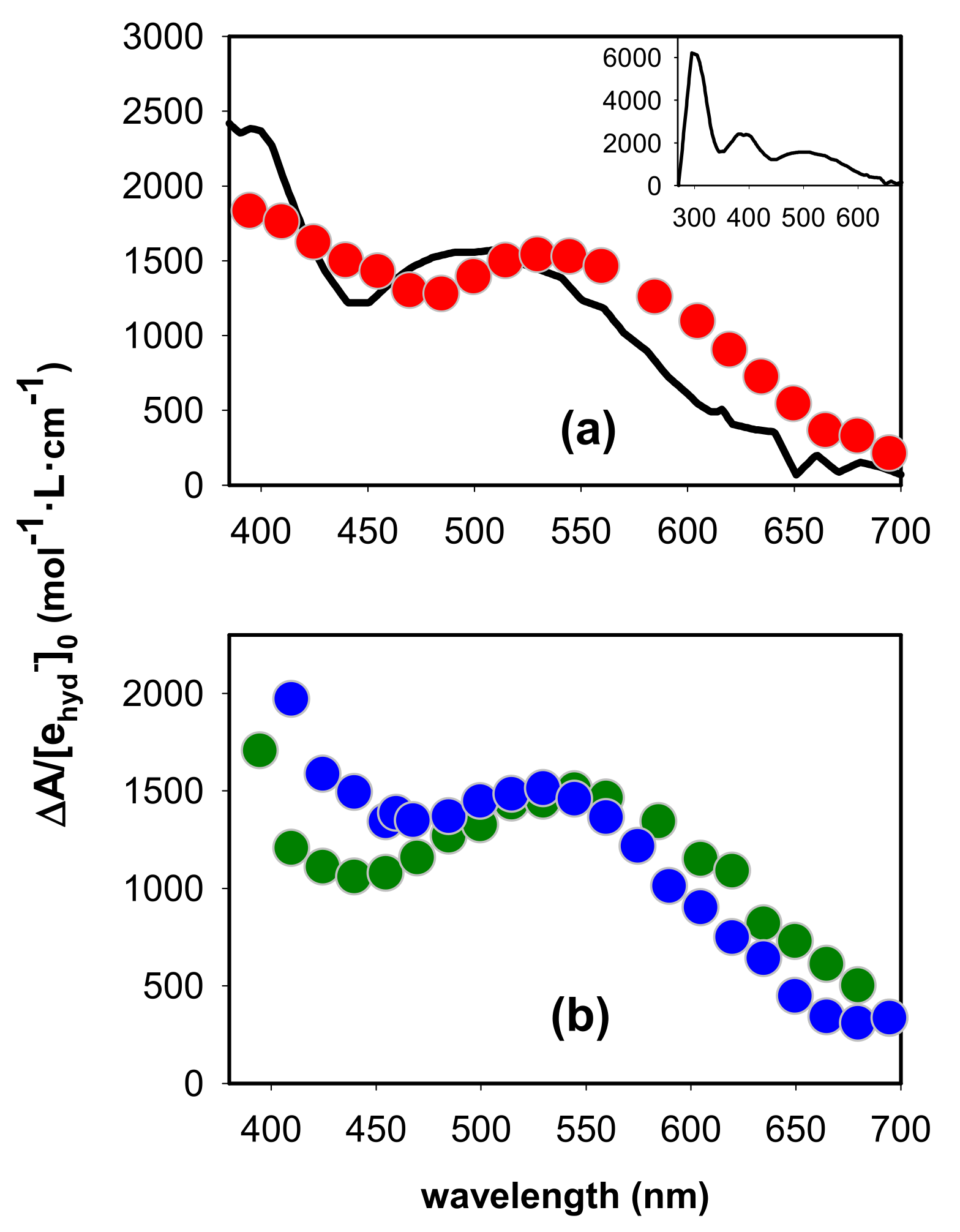
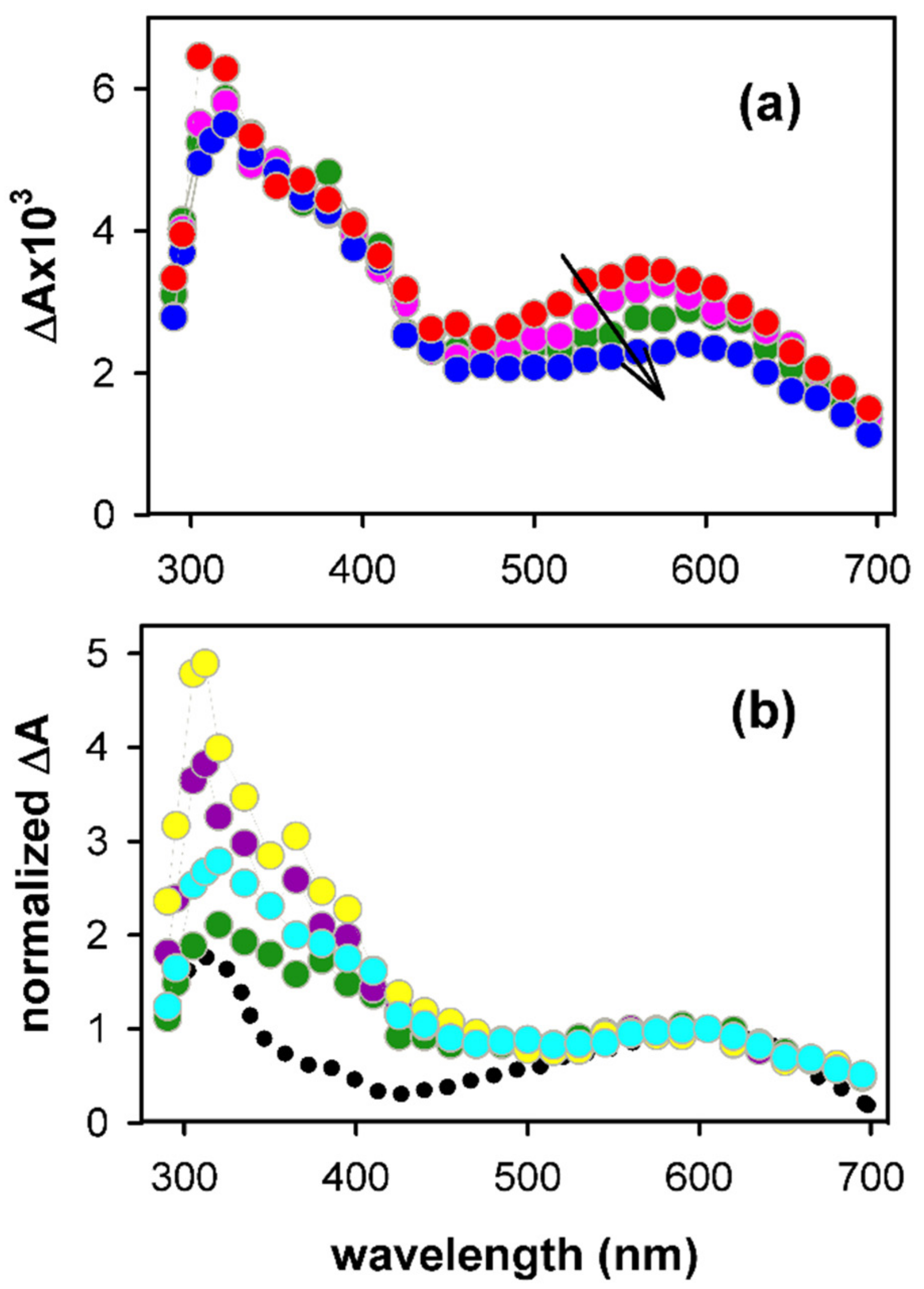
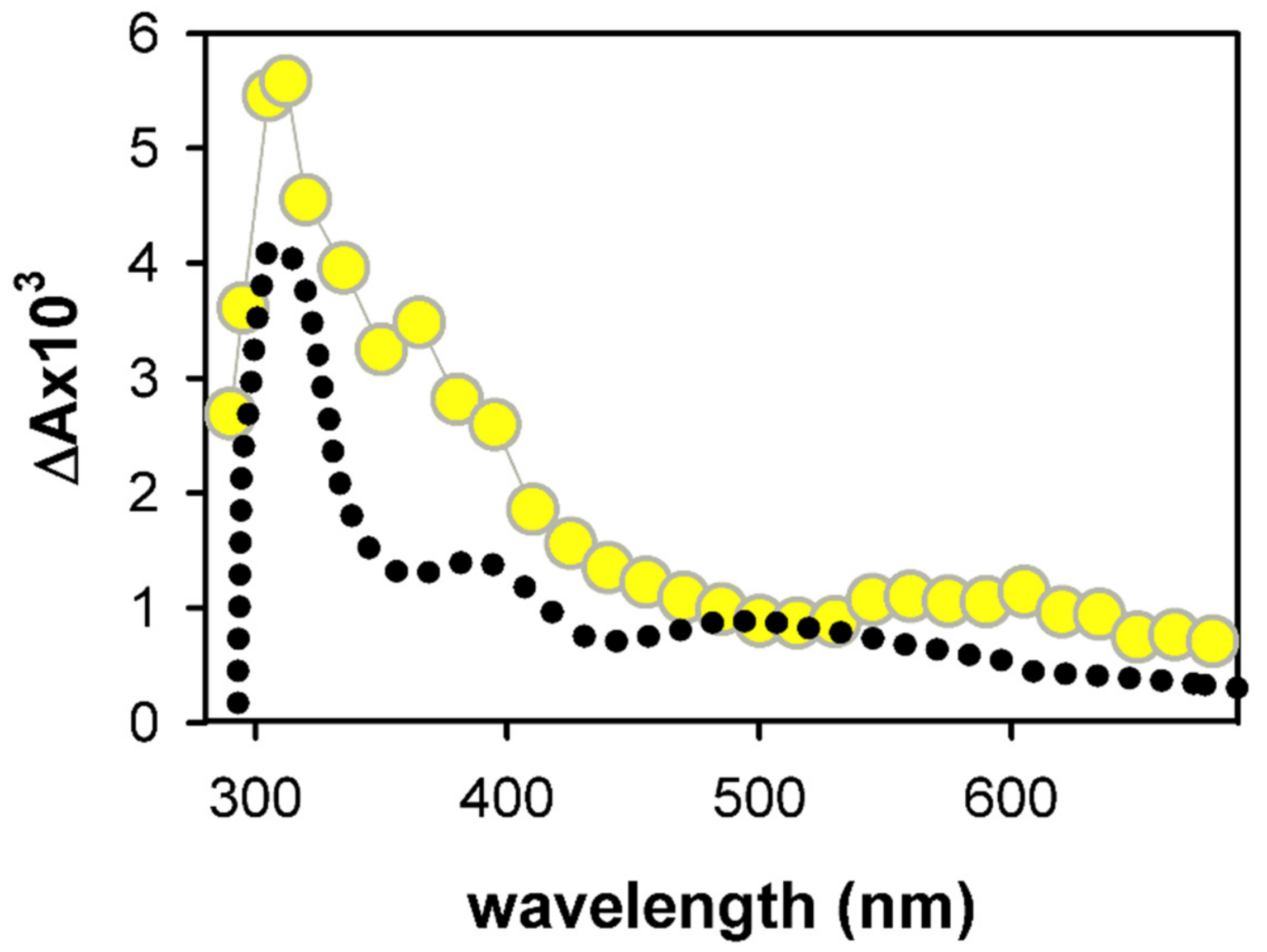
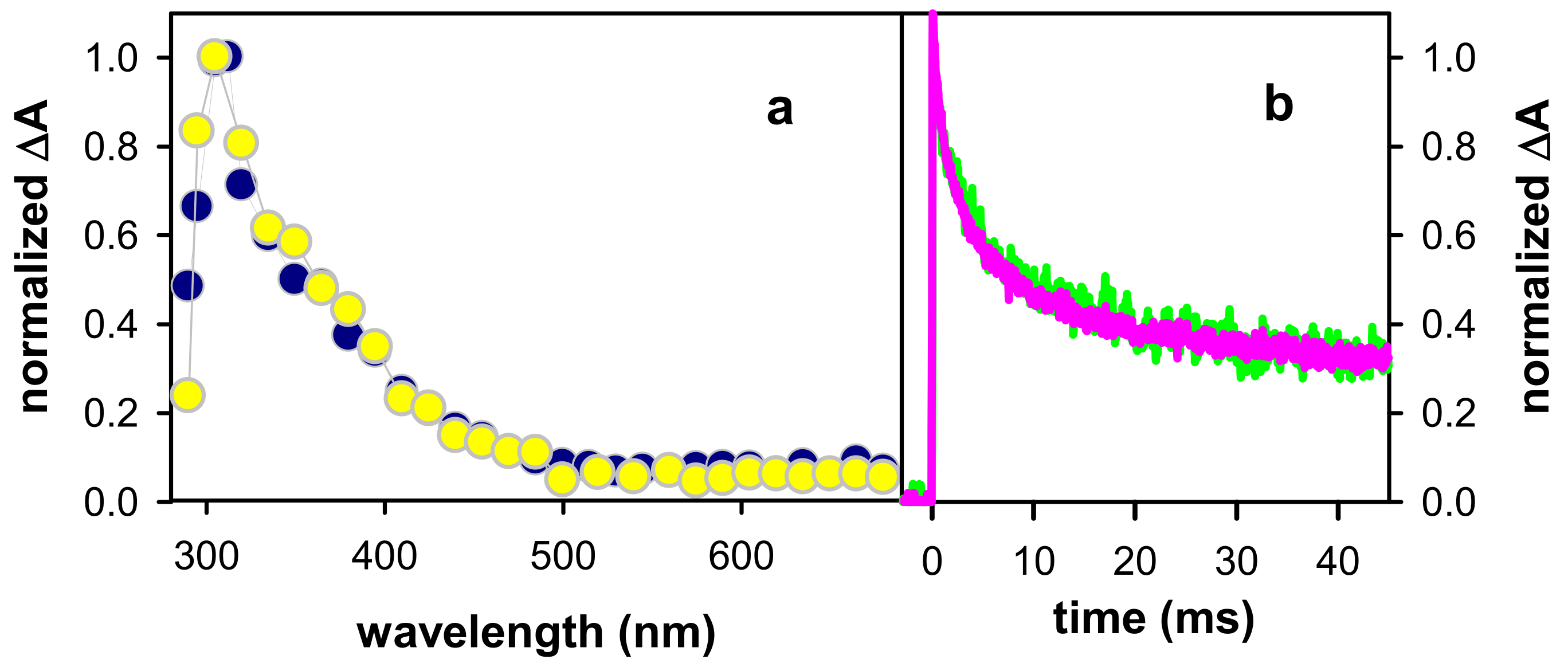
2.1. Spectral Properties
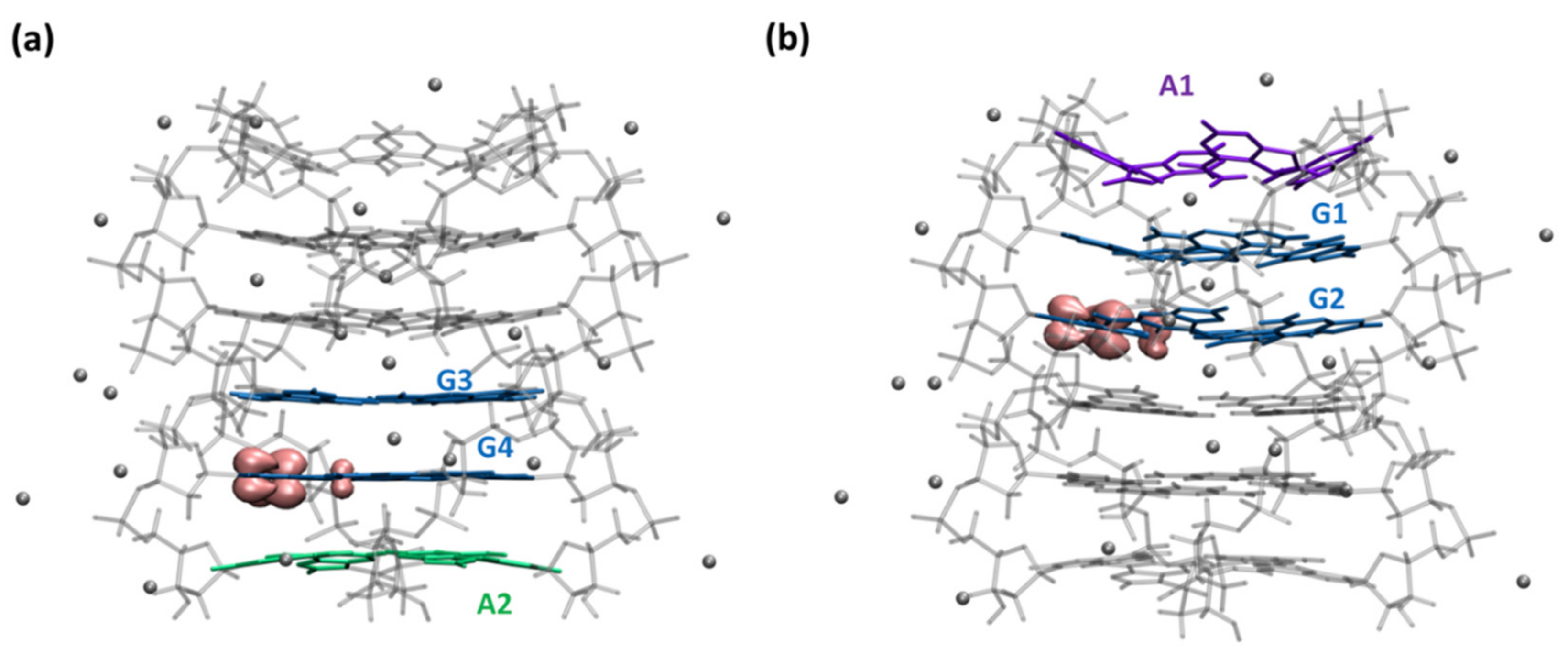
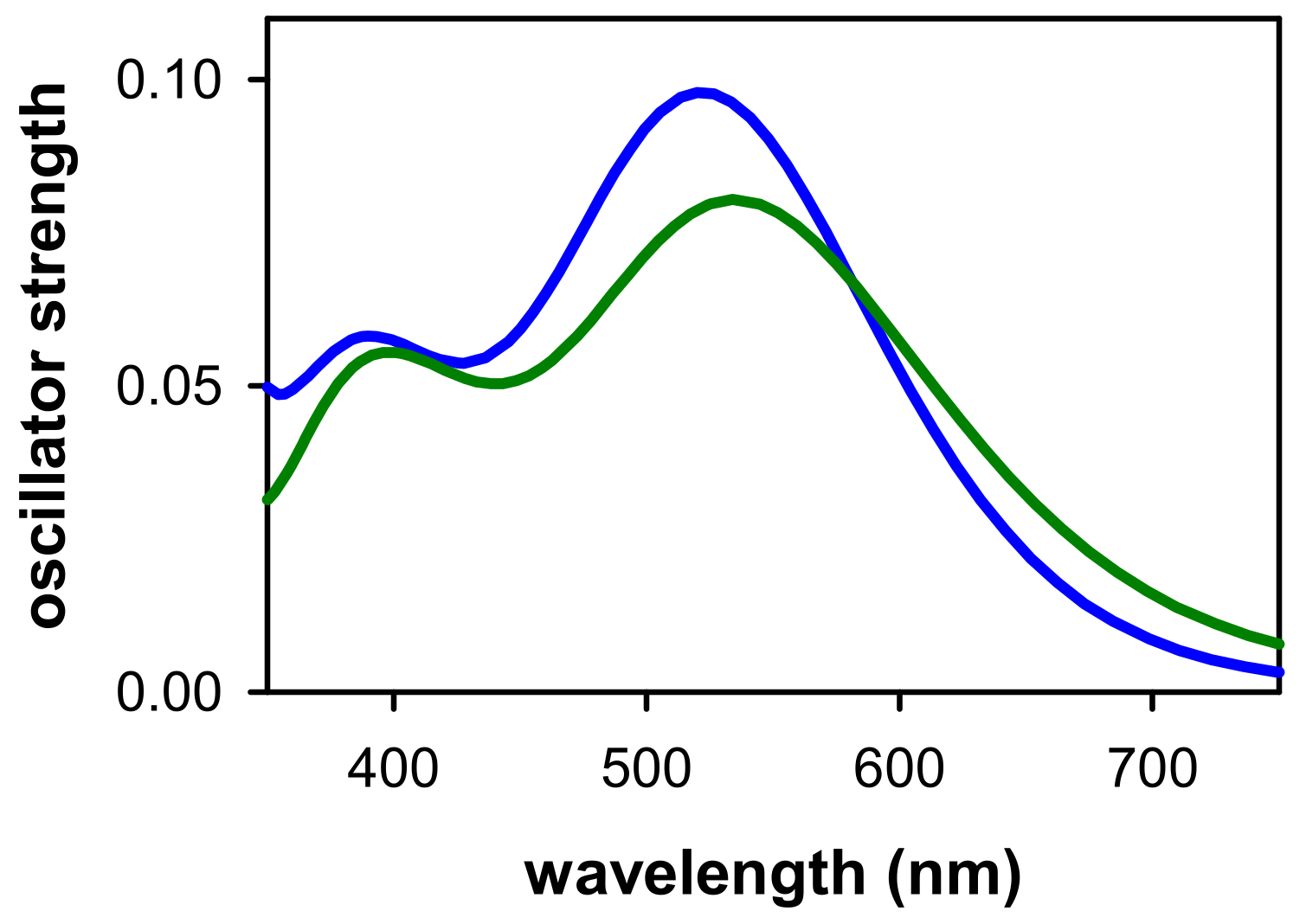
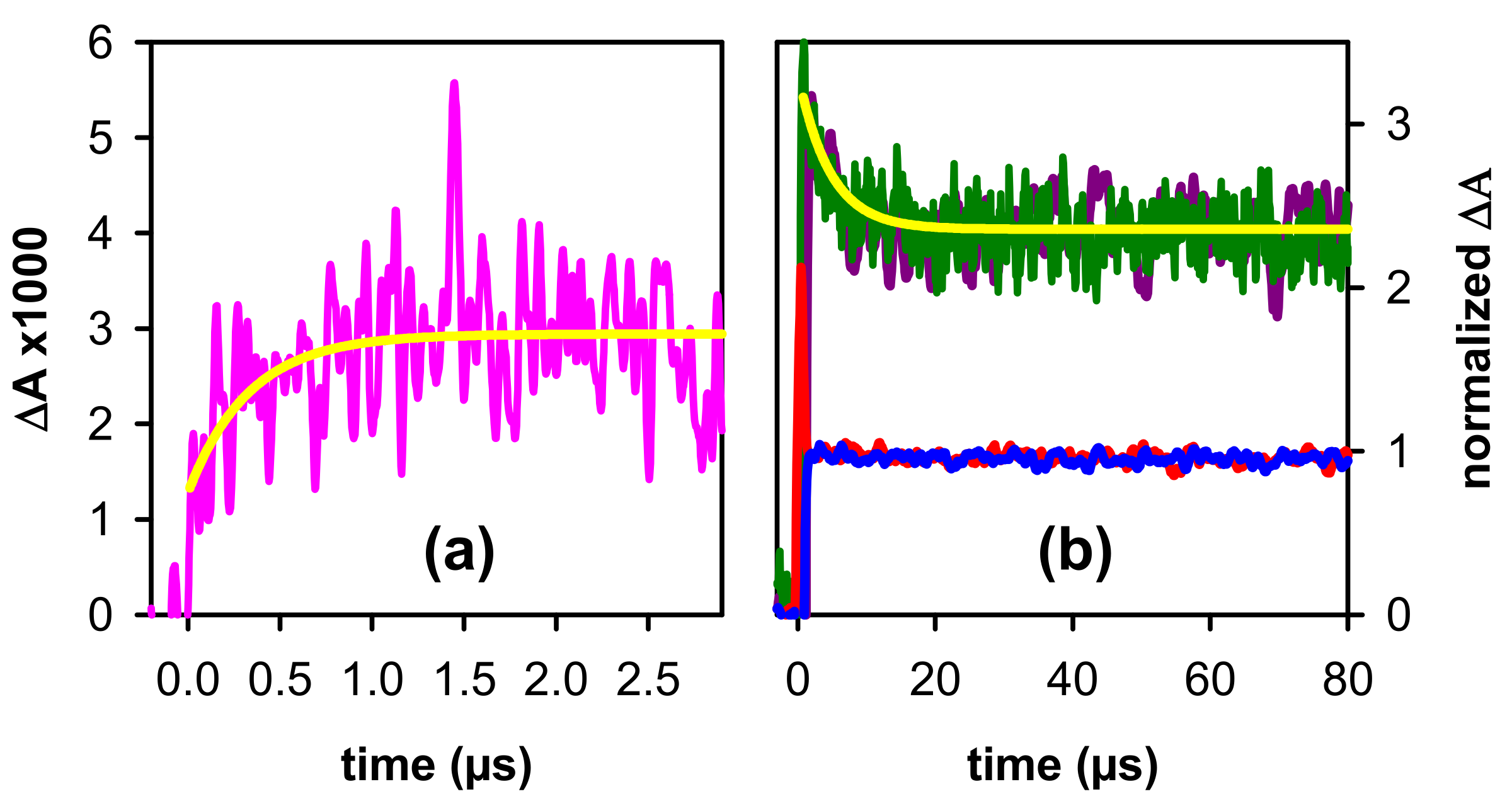
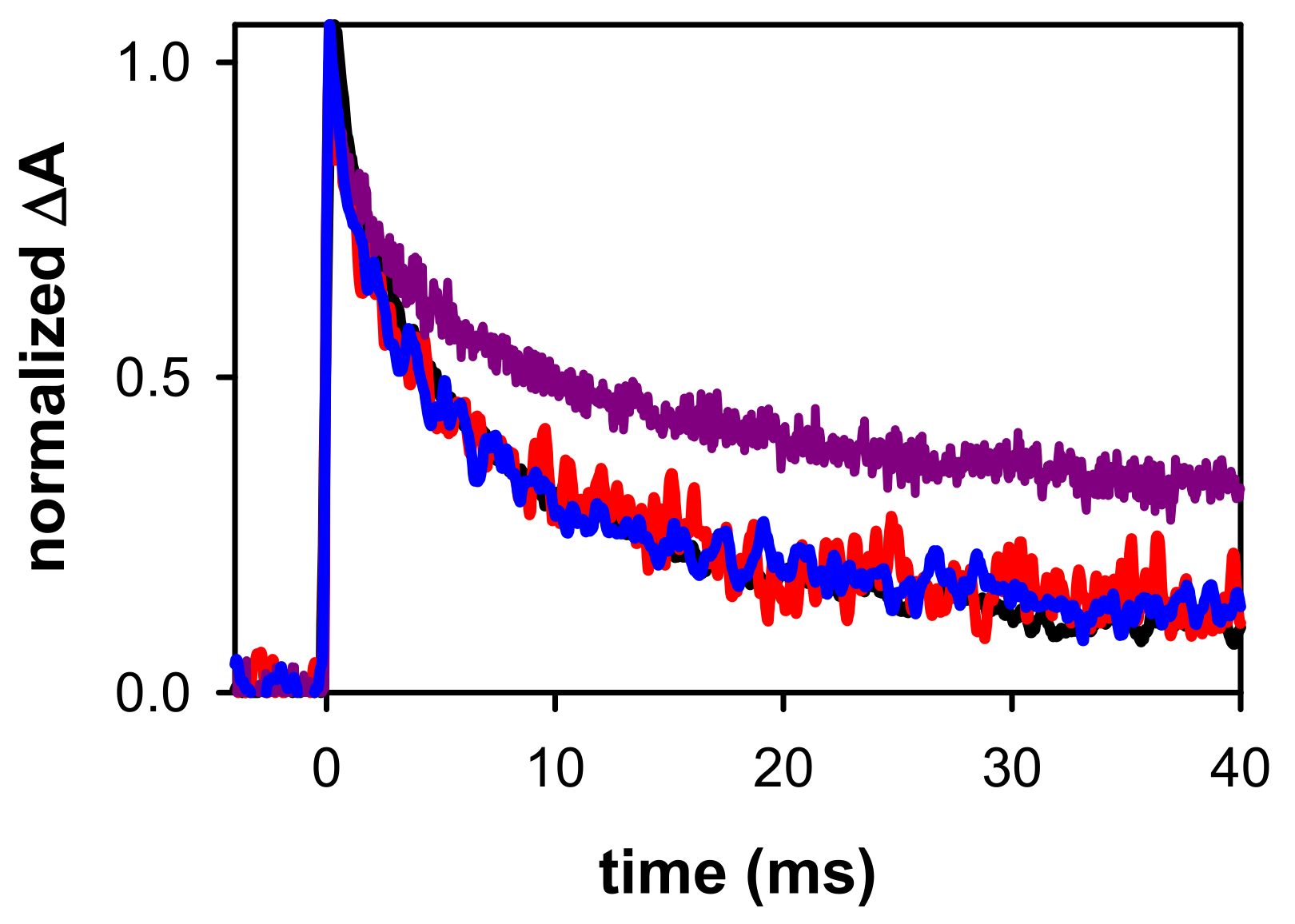
2.2. Dynamical Properties
3. Materials and Methods
3.1. Materials
3.2. Transient Absorption Spectroscopy
3.3. QM/MM Calculations
4. Main Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, D.B.; Holmlin, R.E.; Barton, J.K. Oxidative DNA damage through long-range electron transfer. Nature 1996, 382, 731–735. [Google Scholar] [CrossRef]
- Douki, T.; Angelov, D.; Cadet, J. UV laser photolysis of DNA: Effect of duplex stability on charge-transfer efficiency. J. Am. Chem. Soc. 2001, 123, 11360–11366. [Google Scholar] [CrossRef]
- Stemp, E.D.A.; Arkin, M.R.; Barton, J.K. Oxidation of guanine in DNA by Ru(phen)(2)(dppz)(3+) using the flash-quench technique. J. Am. Chem. Soc. 1997, 119, 2921–2925. [Google Scholar] [CrossRef]
- Meggers, E.; Michel-Beyerle, M.E.; Giese, B. Sequence dependent long range hole transport in DNA. J. Am. Chem. Soc. 1998, 120, 12950–12955. [Google Scholar] [CrossRef]
- Saito, I.; Nakamura, T.; Nakatani, K.; Yoshioka, Y.; Yamaguchi, K.; Sugiyama, H. Mapping of the hot spots for DNA damage by one-electron oxidation: Efficacy of GG doublets and GGG triplets as a trap in long-range hole migration. J. Am. Chem. Soc. 1998, 120, 12686–12687. [Google Scholar] [CrossRef]
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.D.; Letsinger, R.L.; Wasielewski, M.R. Dynamics of photoinduced charge transfer and hole transport in synthetic DNA hairpins. Acc. Chem. Res. 2001, 34, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole transfer kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Ma, J.; Marignier, J.L.; Pernot, P.; Houee-Levin, C.; Kumar, A.; Sevilla, M.D.; Adhikary, A.; Mostafavi, M. Direct observation of the oxidation of DNA bases by phosphate radicals formed under radiation: A model of the backbone-to-base hole transfer. Phys. Chem. Chem. Phys. 2018, 20, 14927–14937. [Google Scholar] [CrossRef]
- Palecek, E.; Bartosik, M. Electrochemistry of nucleic acids. Chem. Rev. 2012, 112, 3427–3481. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Kizhuveetil, U.; Omer, S.; Karunagaran, D.; Suraishkumar, G.K. Improved redox anti-cancer treatment efficacy through reactive species rhythm manipulation. Sci. Rep. 2020, 10, 1588. [Google Scholar] [CrossRef]
- Mergny, J.L.; Sen, D. DNA Quadruple helices in nanotechnology. Chem. Rev. 2019, 119, 6290–6325. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.I.; Stern, A.; Rotem, D.; Borovok, N.; Eidelshtein, G.; Migliore, A.; Penzo, E.; Wind, S.J.; Di Felice, R.; Skourtis, S.S.; et al. Long-range charge transport in single G-quadruplex DNA molecules. Nat. Nanotechnol. 2014, 9, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Delaney, S.; Barton, J.K. Charge transport in DNA duplex/quadruplex conjugates. Biochemistry 2003, 42, 14159–14165. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, J.; Tanaka, A.; Park, M.J.; Jang, Y.J.; Fujitsuka, M.; Kim, S.K.; Majima, T. Hole trapping of G-quartets in a G-quadruplex. Angew. Chem. Int. Ed. 2013, 52, 1134–1138. [Google Scholar] [CrossRef]
- Thazhathveetil, A.K.; Harris, M.A.; Young, R.M.; Wasielewski, M.R.; Lewis, F.D. Efficient charge transport via DNA G-quadruplexes. J. Am. Chem. Soc. 2017, 139, 1730–1733. [Google Scholar] [CrossRef]
- Liu, S.P.; Weisbrod, S.H.; Tang, Z.; Marx, A.; Scheer, E.; Erbe, A. Direct measurement of electrical transport through G-quadruplex DNA with mechanically controllable break junction electrodes. Angew. Chem. Int. Ed. 2010, 49, 3313–3316. [Google Scholar] [CrossRef]
- Sha, R.J.; Xiang, L.M.; Liu, C.R.; Balaeff, A.; Zhang, Y.Q.; Zhang, P.; Li, Y.Q.; Beratan, D.N.; Tao, N.J.; Seeman, N.C. Charge splitters and charge transport junctions based on guanine quadruplexes. Nat. Nanotechnol. 2018, 13, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liang, S.D. Topological effects of charge transfer in telomere G-quadruplex: Mechanism on telomerase activation and inhibition. Inter. J. Mod. Phys. B 2013, 27, 1350001. [Google Scholar] [CrossRef]
- Calzolari, A.; Di Felice, R.; Molinari, E.; Garbesi, A. Electron channels in biomolecular nanowires. J. Phys. Chem. B 2004, 108, 2509–2515. [Google Scholar] [CrossRef]
- Lech, C.J.; Anh Tuan, P.; Michel-Beyerle, M.-E.; Voityuk, A.A. Electron-hole transfer in G-quadruplexes with different tetrad stacking geometries: A combined QM and MD study. J. Phys. Chem. B 2013, 117, 9851–9856. [Google Scholar] [CrossRef]
- Kang, D.W.; Sun, M.L.; Zuo, Z.W.; Wang, H.X.; Lv, S.J.; Li, X.Z.; Li, L.B. Charge transport and magnetoresistance of G4-DNA molecular device modulated by counter ions and dephasing effect. Phys. Lett. A 2016, 380, 977–982. [Google Scholar] [CrossRef]
- Sun, W.M.; Varsano, D.; Di Felice, R. Effects of G-quadruplex topology on electronic transfer integrals. Nanomaterials 2016, 6, 6100184. [Google Scholar] [CrossRef]
- Ravindranath, R.; Mondal, P.; Gillet, N. Radical cation transfer in a guanine pair: An insight to the G-quadruplex structure role using constrained DFT/MM. Theor. Chem. Acc. 2021, 140, 89. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of low-energy UV Radiation by human telomere g-quadruplexes generates long-lived guanine radical cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Balanikas, E.; Martinez-Fernandez, L.; Baldacchino, G.; Douki, T.; Improta, R.; Markovitsi, D. Radicals generated in tetramolecular guanine quadruplexes by photo-ionization: Spectral and dynamical features. J. Phys. Chem. B 2019, 123, 4950–4957. [Google Scholar] [CrossRef] [PubMed]
- Marguet, S.; Markovitsi, D.; Talbot, F. One and two photon ionization of DNA single and double helices studied by laser flash photolysis at 266 nm. J. Phys. Chem. B 2006, 110, 11037–11039. [Google Scholar] [CrossRef][Green Version]
- Balanikas, E.; Banyasz, A.; Douki, T.; Baldacchino, G.; Markovitsi, D. Guanine radicals induced in DNA by low-energy photoionization. Acc. Chem. Res. 2020, 53, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Balanikas, E.; Markovitsi, D. DNA photoionization: From high to low energies. In DNA Photodamage; Improta, R., Douki, T., Eds.; RSC: Cambridge, UK, 2021; pp. 37–54. [Google Scholar]
- Marguet, S.; Markovitsi, D. Time-resolved study of thymine dimer formation. J. Am. Chem. Soc. 2005, 127, 5780–5781. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Ketola, T.; Muñoz-Losa, A.; Esposito, L.; Markovitsi, D.; Improta, R. Excited state pathways leading to formation of adenine dimers. J. Phys. Chem. Lett. 2016, 7, 2020–2023. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Ionization of purine nucleosides and nucleotides and their components by 193-nm laser photolysis in aqueous solution: Model studies for oxidative damage of DNA. J. Am. Chem. Soc. 1992, 114, 699–704. [Google Scholar] [CrossRef]
- Wu, L.D.; Liu, K.H.; Jie, J.L.; Song, D.; Su, H.M. Direct observation of guanine radical cation deprotonation in G-quadruplex DNA. J. Am. Chem. Soc. 2015, 137, 259–266. [Google Scholar] [CrossRef]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Deprotonation dynamics of guanine radical cations. Photochem. Photobiol. 2021, 13, 540. [Google Scholar] [CrossRef]
- Balanikas, E.; Martinez-Fernadez, L.; Improta, R.; Podbevsek, P.; Baldacchino, G.; Markovitsi, D. The structural duality of nucleobases in guanine quadruplexes controls their low-energy photoionization. J. Phys. Chem. Lett. 2021, 12, 8309–8313. [Google Scholar] [CrossRef] [PubMed]
- Pluharova, E.; Slavicek, P.; Jungwirth, P. Modeling photoionization of aqueous DNA and its components. Acc. Chem. Res. 2015, 48, 1209–1217. [Google Scholar] [CrossRef]
- Khanduri, D.; Adhikary, A.; Sevilla, M.D. Highly oxidizing excited states of one-electron-oxidized guanine in DNA: Wavelength and pH dependence. J. Am. Chem. Soc. 2011, 133, 4527–4537. [Google Scholar] [CrossRef] [PubMed]
- Candeias, L.P.; Steenken, S. Stucture and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Sket, P.; Plavec, J. Tetramolecular DNA quadruplexes in solution: Insights into structural diversity and cation movement. J. Am. Chem. Soc. 2010, 132, 12724–12732. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Banyasz, A.; Markovitsi, D.; Improta, I. Topology controls the electronic absorption delocalization of electron hole in guanine quadruplexes. Chem. Eur. J. 2018, 24, 15185–15189. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. Density functional theory studies of the extent of hole delocalization in one-electron oxidized adenine and guanine base stacks. J. Phys. Chem. B 2011, 115, 4990–5000. [Google Scholar] [CrossRef]
- Adhikary, A.; Malkhasian, A.Y.S.; Collins, S.; Koppen, J.; Becker, D.; Sevilla, M.D. UVA-visible photo-excitation of guanine radical cations produces sugar radicals in DNA and model structures. Nucl. Ac. Res. 2005, 33, 5553–5564. [Google Scholar] [CrossRef]
- Adhikary, A.; Kumar, A.; Becker, D.; Sevilla, M.D. The guanine cation radical: Investigation of deprotonation states by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Li, Y.; Li, S.; Zhang, M.; Zhou, Z. Collision-induced dissociation (CID) of guanine radical cation in the gas phase: An experimental and computational study. Phys. Chem. Chem. Phys. 2010, 12, 4667–4677. [Google Scholar] [CrossRef] [PubMed]
- Behmand, B.; Balanikas, E.; Martinez-Fernandez, L.; Improta, R.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Potassium ions enhance guanine radical generation upon absorption of low-energy photons by G-quadruplexes and modify their reactivity. J. Phys. Chem. Lett. 2020, 11, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Esposito, L.; Improta, R. Studying the excited electronic states of guanine rich DNA quadruplexes by quantum mechanical methods: Main achievements and perspectives. Photochem. Photobiol. Sci. 2020, 19, 436–444. [Google Scholar] [CrossRef]
- Santoro, F.; Barone, V.; Lami, A.; Improta, R. The excited electronic states of adenine-guanine stacked dimers in aqueous solution: A PCM/TD-DFT study. Phys. Chem. Chem. Phys. 2010, 12, 4934–4948. [Google Scholar] [CrossRef] [PubMed]
- Aquino, A.J.A.; Nachtigallova, D.; Hobza, P.; Truhlar, D.G.; Hattig, C.; Lischka, H. The charge-transfer states in a stacked nucleobase dimer complex: A benchmark study. J. Comput. Chem. 2011, 32, 1217–1227. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Improta, R. Novel adenine/thymine photodimerization channels mapped by PCM/TD-DFT calculations on dApT and TpdA dinucleotides. Photochem. Photobiol. Sci 2017, 16, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Changenet-Barret, P.; Improta, R.; Vayá, I.; Gustavsson, T.; Kotlyar, A.B.; Zikich, D.; Šket, P.; Plavec, J.; Markovitsi, D. Cation effect on the electronic excited states of guanine nanostructures studied by time-resolved fluorescence spectroscopy. J. Phys. Chem. C 2012, 116, 14682–14689. [Google Scholar] [CrossRef]
- Improta, R. Quantum mechanical calculations unveil the structure and properties of the absorbing and emitting excited electronic states of guanine quadruplex. Chem. Eur. J. 2014, 20, 8106–8115. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Changenet, P.; Banyasz, A.; Gustavsson, T.; Markovitsi, D.; Improta, R. A Comprehensive study of guanine excited state relaxation and photoreactivity in G-quadruplexes. J. Phys. Chem. Lett. 2019, 10, 6873–6877. [Google Scholar] [CrossRef]
- Chatgilialoglu, C. The two faces of the guanyl radical: Molecular context and behavior. Molecules 2021, 26, 3511. [Google Scholar] [CrossRef]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair; Springer: Berlin/Heildelberg, Germany, 2006; p. 523. [Google Scholar]
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Guanine radicals generated in telomeric G-quadruplexes by direct absorption of low-energy UV photons: Effect of potassium ions. Molecules 2020, 25, 2094. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Kumar, A.; Muroya, Y.; Yamashita, S.; Sakurai, T.; Denisov, S.A.; Sevilla, M.D.; Adhikary, A.; Seki, S.; Mostafavi, M. Observation of dissociative quasi-free electron attachment to nucleoside via excited anion radical in solution. Nat. Commun. 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem. Res.Toxicol. 2013, 26, 593–607. [Google Scholar] [CrossRef]
- Bielskute, S.; Plavec, J.; Podbevsek, P. Impact of oxidative lesions on the human telomeric G-quadruplex. J. Am. Chem. Soc. 2019, 141, 2594–2603. [Google Scholar] [CrossRef]
- Dapprich, S.; Komaromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part, I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct.-Theochem 1999, 461, 1–21. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A 2nd generation force-field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum-A direct utilization of abinitio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Borrego-Varillas, R.; Cerullo, G.; Markovitsi, D. Exciton trapping dynamics in DNA multimers. J. Phys. Chem. Lett. 2019, 10, 1639–1643. [Google Scholar] [CrossRef]
- Ma, J.; Wang, F.; Denisov, S.A.; Adhikary, A.; Mostafavi, M. Reactivity of prehydrated electrons toward nucleobases and nucleotides in aqueous solution. Sci. Adv. 2017, 3, e1701669. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balanikas, E.; Martinez-Fernandez, L.; Baldacchino, G.; Markovitsi, D. Electron Holes in G-Quadruplexes: The Role of Adenine Ending Groups. Int. J. Mol. Sci. 2021, 22, 13436. https://doi.org/10.3390/ijms222413436
Balanikas E, Martinez-Fernandez L, Baldacchino G, Markovitsi D. Electron Holes in G-Quadruplexes: The Role of Adenine Ending Groups. International Journal of Molecular Sciences. 2021; 22(24):13436. https://doi.org/10.3390/ijms222413436
Chicago/Turabian StyleBalanikas, Evangelos, Lara Martinez-Fernandez, Gérard Baldacchino, and Dimitra Markovitsi. 2021. "Electron Holes in G-Quadruplexes: The Role of Adenine Ending Groups" International Journal of Molecular Sciences 22, no. 24: 13436. https://doi.org/10.3390/ijms222413436
APA StyleBalanikas, E., Martinez-Fernandez, L., Baldacchino, G., & Markovitsi, D. (2021). Electron Holes in G-Quadruplexes: The Role of Adenine Ending Groups. International Journal of Molecular Sciences, 22(24), 13436. https://doi.org/10.3390/ijms222413436