
Environment-Sensitive Fluorescent Labelling of Peptides by Luciferin Analogues

 , ,
, ,  , ,
, ,  , , , , , and
, , , , , and 
Abstract
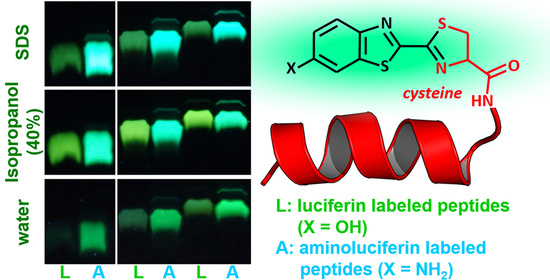
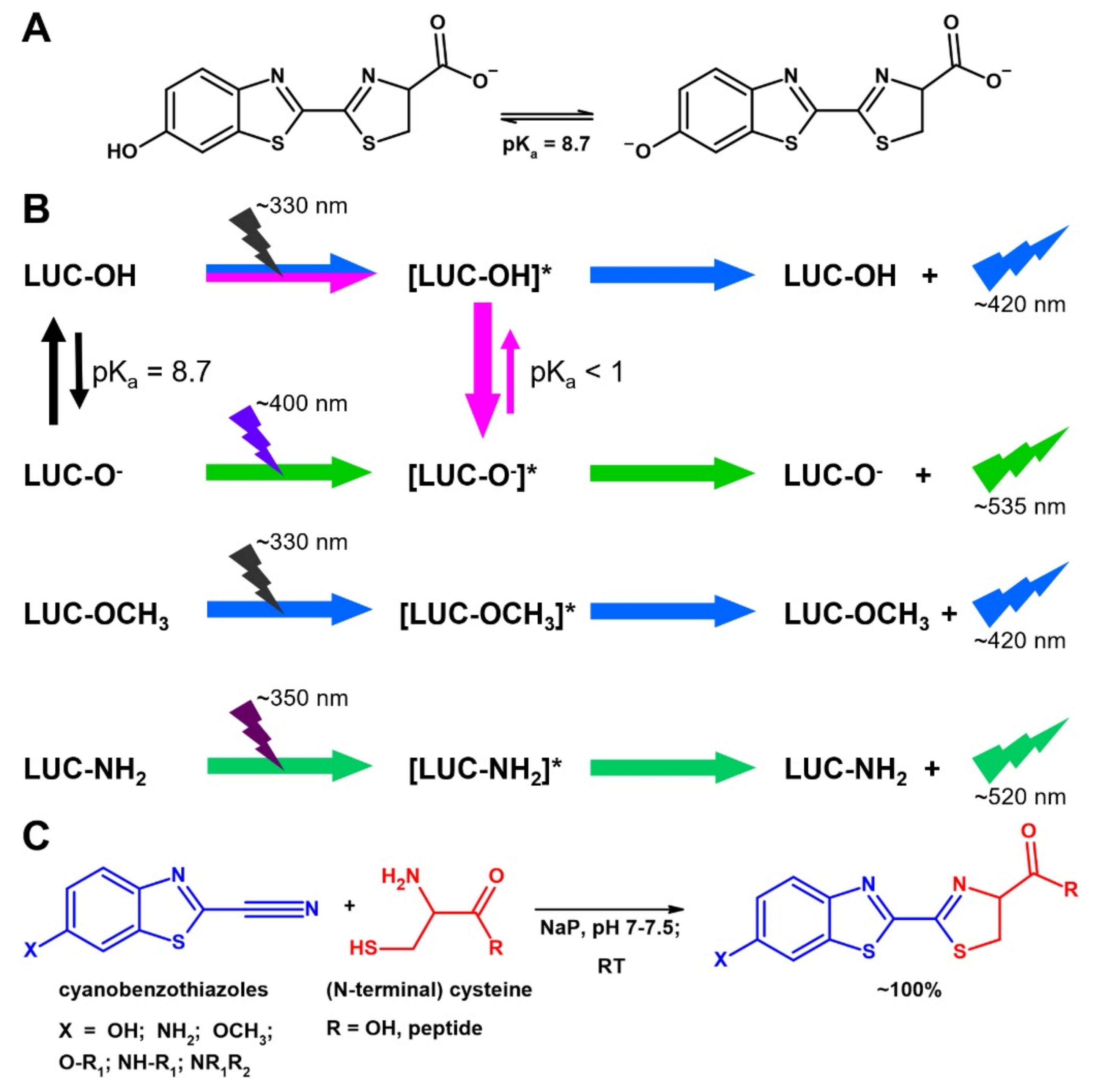
1. Introduction
2. Results
2.1. Preparation and Labeling of the Peptides
2.2. Stability and Biological Activity of Luc-Labeled Peptides
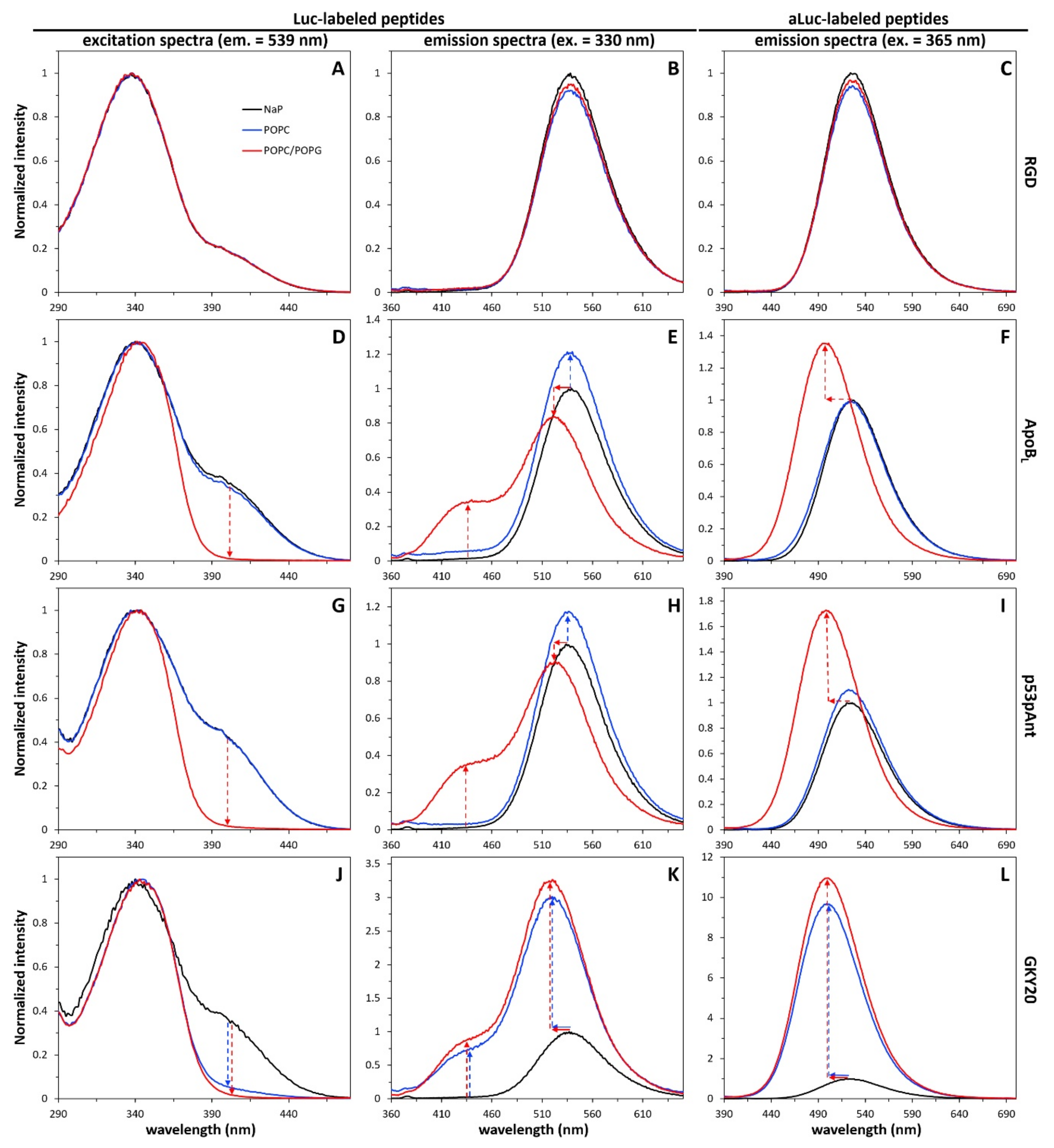
2.3. Response of the Labeled Peptides to Solvent Polarity Changes
2.4. Response of Luc-Labeled Peptides to pH
2.5. Interaction of Labeled Peptides with Liposomes
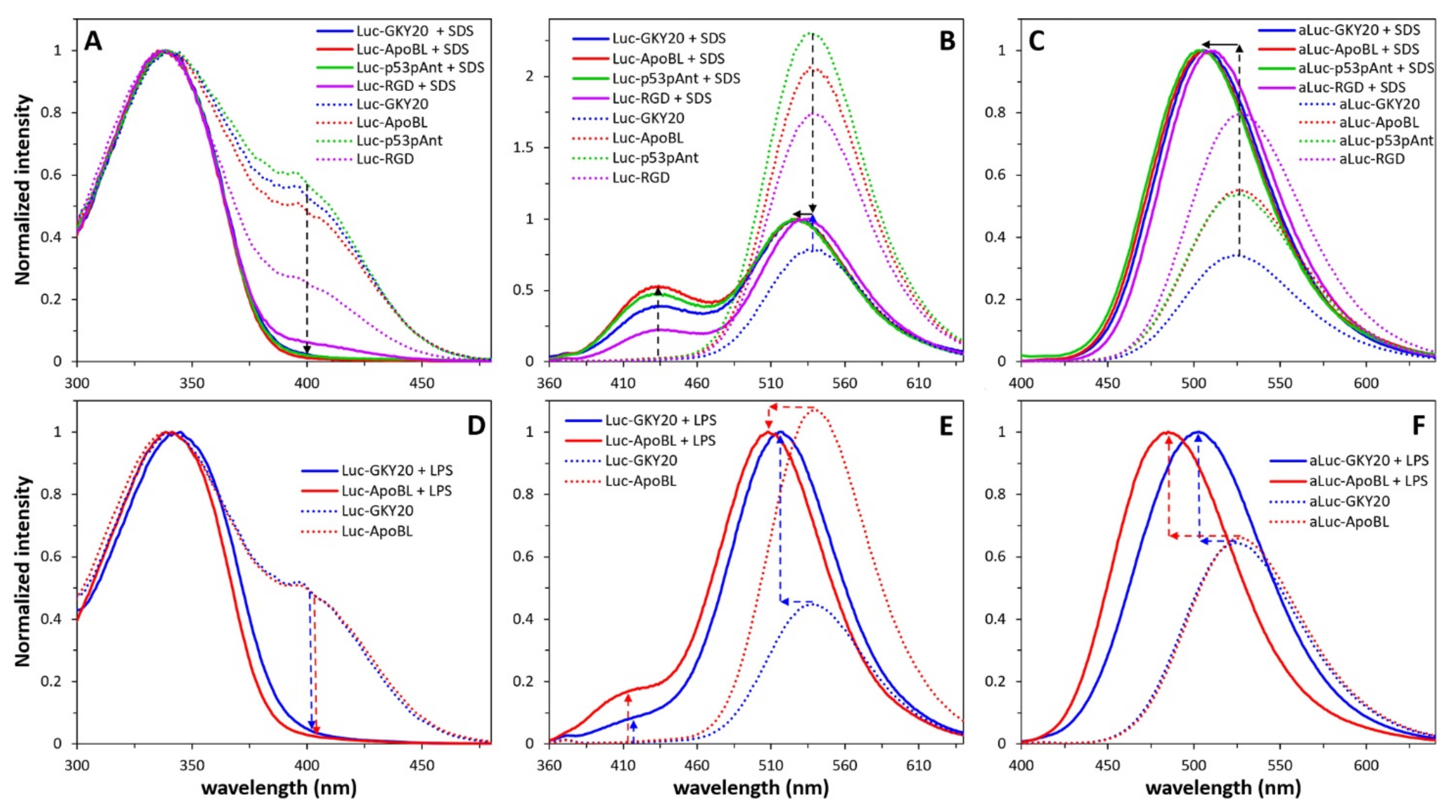
2.6. Interaction of Labeled CAMPs with SDS and LPS Micelles
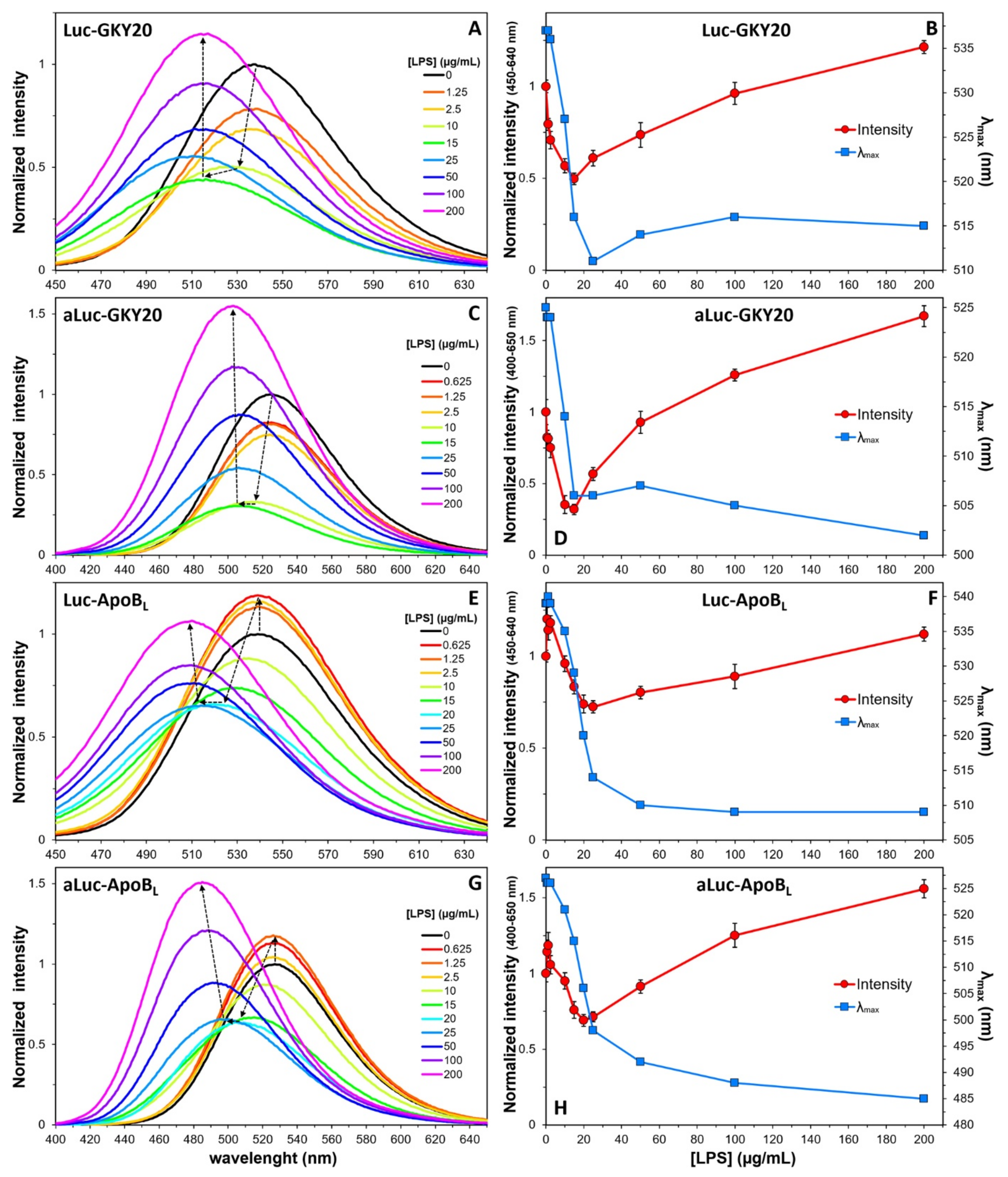
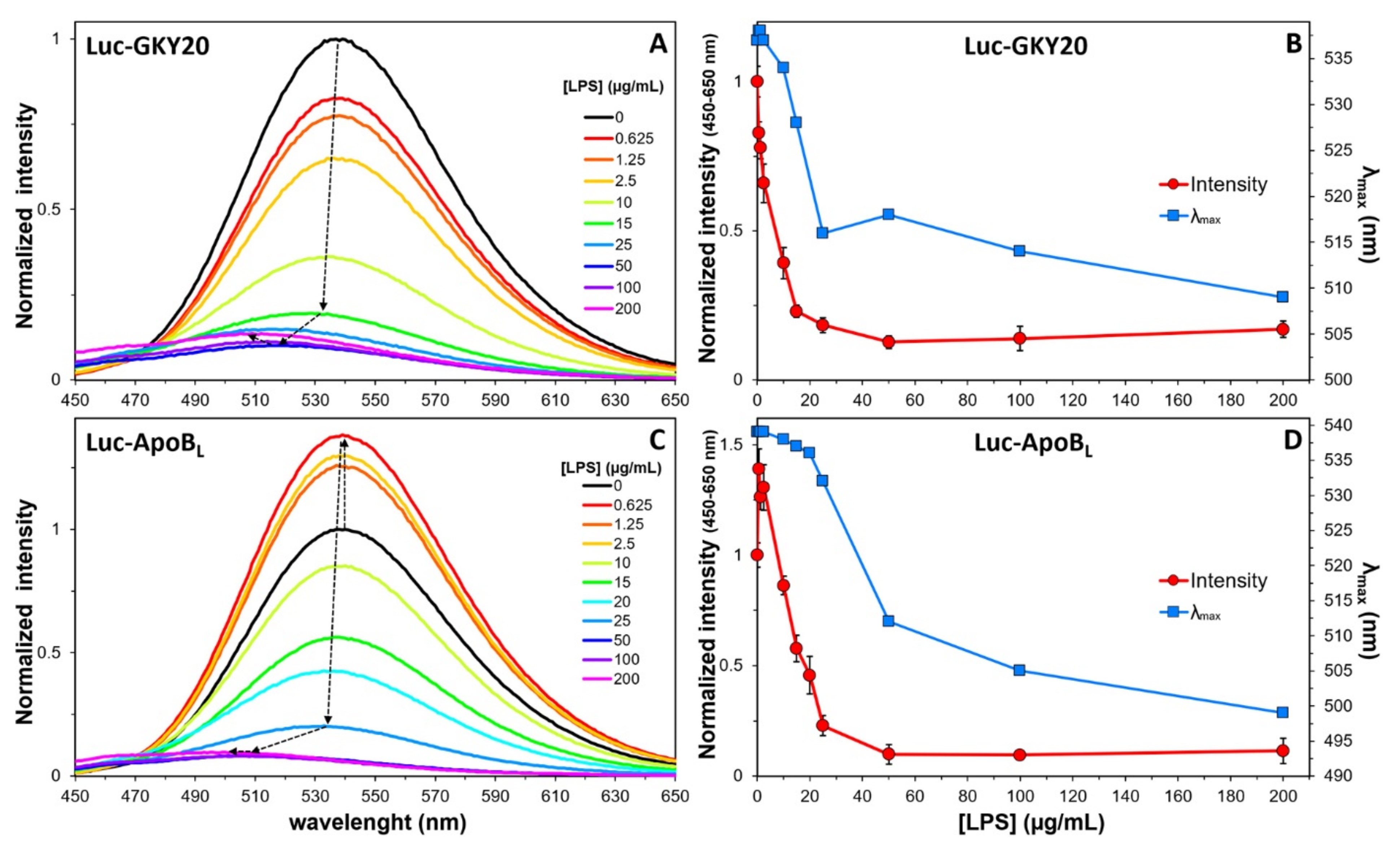
2.7. Interaction of Labeled CAMPs with Non-Micellar LPS
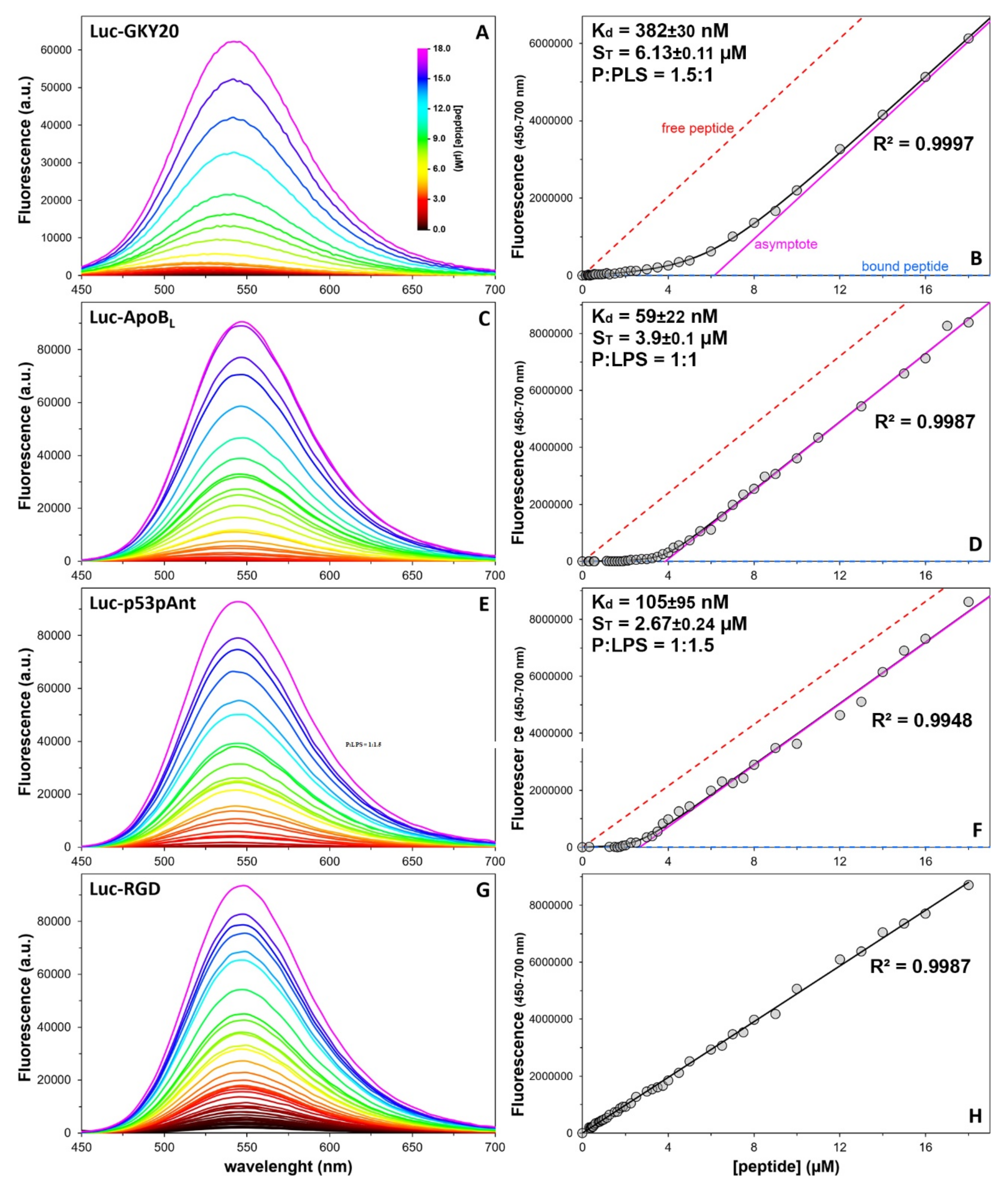
2.8. Quantitative Analysis of the Peptide/LPS Interaction
2.9. Interaction of Labeled CAMPs with E. coli Cells
2.10. Confocal Laser Scanning Microscopy
3. Discussion
4. Materials and Methods
4.1. Materials and General Methods
4.2. Steady-State Fluorescence Spectroscopy in Water/Organic Solvent Mixtures and SDS
4.3. Steady-State Fluorescence Spectroscopy in the Presence of Liposomes
4.4. Quantum-Yield Determination
4.5. pKa Determination of Luc-Labeled Peptides
4.6. Interaction of Labeled CAMPs with LPS
4.7. Interaction of Labeled CAMPs with E. coli Cells
4.8. Kinetic Analysis
4.9. Microscopy Analysis of E. coli Cells Treated with the Labeled Peptides
4.10. Interaction of aLuc-p53pAnt and aLuc-RGD with HeLa and HaCaT Cells
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Klymchenko, A.S. Solvatochromic and Fluorogenic Dyes as Environment-Sensitive Probes: Design and Biological Applications. Acc. Chem. Res. 2017, 50, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Burgess, K. Fluorescent Indicators for Intracellular pH. Chem. Rev. 2010, 110, 2709–2728. [Google Scholar] [CrossRef] [PubMed]
- Loving, G.S.; Sainlos, M.; Imperiali, B. Monitoring protein interactions and dynamics with solvatochromic fluorophores. Trends Biotechnol. 2010, 28, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Donadio, G.; Di Martino, R.; Oliva, R.; Petraccone, L.; Del Vecchio, P.; Di Luccia, B.; Ricca, E.; Isticato, R.; Di Donato, A.; Notomista, E. A new peptide-based fluorescent probe selective for zinc(II) and copper(II). J. Mater. Chem. B 2016, 4, 6979–6988. [Google Scholar] [CrossRef]
- Li, S.; Ruan, Z.; Zhang, H.; Xu, H. Recent achievements of bioluminescence imaging based on firefly luciferin-luciferase system. Eur. J. Med. Chem. 2021, 211, 113111. [Google Scholar] [CrossRef]
- Morton, R.A.; Hopkins, T.A.; Seliger, H.H. Spectroscopic properties of firefly luciferin and related compounds; an approach to product emission. Biochemistry 1969, 8, 1598–1607. [Google Scholar] [CrossRef]
- Ando, Y.; Akiyama, H. PH-dependent fluorescence spectra, lifetimes, and quantum yields of firefly-luciferin aqueous solutions studied by selective-excitation fluorescence spectroscopy. Jpn. J. Appl. Phys. 2010, 49, 117002. [Google Scholar] [CrossRef]
- Presiado, I.; Erez, Y.; Huppert, D. Excited-state intermolecular proton transfer of the firefly’s chromophore d-luciferin. 2. water-methanol mixtures. J. Phys. Chem. A 2010, 114, 9471–9479. [Google Scholar] [CrossRef]
- Kuchlyan, J.; Banik, D.; Roy, A.; Kundu, N.; Sarkar, N. Excited-state proton transfer dynamics of fireflys chromophore d -luciferin in DMSO-water binary mixture. J. Phys. Chem. B 2014, 118, 13946–13953. [Google Scholar] [CrossRef]
- Vieira, J.; Da Silva, L.P.; Da Silva, J.C.G.E. Advances in the knowledge of light emission by firefly luciferin and oxyluciferin. J. Photochem. Photobiol. B Biol. 2012, 117, 33–39. [Google Scholar] [CrossRef]
- Kakiuchi, M.; Ito, S.; Yamaji, M.; Viviani, V.R.; Maki, S.; Hirano, T. Spectroscopic Properties of Amine-substituted Analogues of Firefly Luciferin and Oxyluciferin. Photochem. Photobiol. 2017, 93, 486–494. [Google Scholar] [CrossRef]
- Zheng, M.; Huang, H.; Zhou, M.; Wang, Y.; Zhang, Y.; Ye, D.; Chen, H.Y. Cysteine-Mediated Intracellular Building of Luciferin to Enhance Probe Retention and Fluorescence Turn-On. Chem. A Eur. J. 2015, 21, 10506–10512. [Google Scholar] [CrossRef]
- Miao, Q.; Li, Q.; Yuan, Q.; Li, L.; Hai, Z.; Liu, S.; Liang, G. Discriminative Fluorescence Sensing of Biothiols in Vitro and in Living Cells. Anal. Chem. 2015, 87, 3460–3466. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, Y.; Shi, H.; Hu, Y.; Feng, L.; Luo, Z.; Zhou, M.; He, J.; Zhou, Z.; Zhang, Y.; et al. Redox-Mediated Disassembly to Build Activatable Trimodal Probe for Molecular Imaging of Biothiols. ACS Nano 2016, 10, 10075–10085. [Google Scholar] [CrossRef]
- Zhao, X.; Lv, G.; Peng, Y.; Liu, Q.; Li, X.; Wang, S.; Li, K.; Qiu, L.; Lin, J. Targeted Delivery of an Activatable Fluorescent Probe for the Detection of Furin Activity in Living Cells. ChemBioChem 2018, 19, 1060–1065. [Google Scholar] [CrossRef]
- Ren, H.; Xiao, F.; Zhan, K.; Kim, Y.P.; Xie, H.; Xia, Z.; Rao, J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. 2009, 48, 9658–9662. [Google Scholar] [CrossRef]
- Chen, K.T.; Ieritano, C.; Seimbille, Y. Early-Stage Incorporation Strategy for Regioselective Labeling of Peptides using the 2-Cyanobenzothiazole/1,2-Aminothiol Bioorthogonal Click Reaction. ChemistryOpen 2018, 7, 256–261. [Google Scholar] [CrossRef]
- Shinde, R.; Perkins, J.; Contag, C.H. Luciferin derivatives for enhanced in vitro and in vivo bioluminescence assays. Biochemistry 2006, 45, 11103–11112. [Google Scholar] [CrossRef]
- Kasetty, G.; Papareddy, P.; Kalle, M.; Rydengård, V.; Mörgelin, M.; Albiger, B.; Malmsten, M.; Schmidtchen, A. Structure-activity studies and therapeutic potential of host defense peptides of human thrombin. Antimicrob. Agents Chemother. 2011, 55, 2880–2890. [Google Scholar] [CrossRef]
- Gaglione, R.; Dell’Olmo, E.; Bosso, A.; Chino, M.; Pane, K.; Ascione, F.; Itri, F.; Caserta, S.; Amoresano, A.; Lombardi, A.; et al. Novel human bioactive peptides identified in Apolipoprotein B: Evaluation of their therapeutic potential. Biochem. Pharmacol. 2017, 130, 34–50. [Google Scholar] [CrossRef]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Pane, K.; Durante, L.; Crescenzi, O.; Cafaro, V.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Izzo, V.; Di Donato, A.; Notomista, E. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: Application to the detection of “cryptic” antimicrobial peptides. J. Theor. Biol. 2017, 419, 254–265. [Google Scholar] [CrossRef]
- Pizzo, E.; Cafaro, V.; Di Donato, A.; Notomista, E. Cryptic Antimicrobial Peptides: Identification Methods and Current Knowledge of their Immunomodulatory Properties. Curr. Pharm. Des. 2018, 24, 1054–1066. [Google Scholar] [CrossRef]
- Dell’Olmo, E.; Gaglione, R.; Cesaro, A.; Cafaro, V.; Teertstra, W.R.; de Cock, H.; Notomista, E.; Haagsman, H.P.; Veldhuizen, E.J.A.; Arciello, A. Host defence peptides identified in human apolipoprotein B as promising antifungal agents. Appl. Microbiol. Biotechnol. 2021, 105, 1953–1964. [Google Scholar] [CrossRef]
- Selivanova, G.; Iotsova, V.; Okan, I.; Fritsche, M.; Ström, M.; Groner, B.; Grafström, R.C.; Wiman, K.G. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 1997, 3, 632–638. [Google Scholar] [CrossRef]
- Li, Y.; Mao, Y.; Rosal, R.V.; Dinnen, R.D.; Williams, A.C.; Brandt-Rauf, P.W.; Fine, R.L. Selective induction of apoptosis through the FADD/Caspase-8 pathway by a p53 C-terminal peptide in human pre-malignant and malignant cells. Int. J. Cancer 2005, 115, 55–64. [Google Scholar] [CrossRef]
- Li, Y.; Rosal, R.V.; Brandt-Rauf, P.W.; Fine, R.L. Correlation between hydrophobic properties and efficiency of carrier-mediated membrane transduction and apoptosis of a p53 C-terminal peptide. Biochem. Biophys. Res. Commun. 2002, 298, 439–449. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef]
- Knetsch, P.A.; Zhai, C.; Rangger, C.; Blatzer, M.; Haas, H.; Kaeopookum, P.; Haubner, R.; Decristoforo, C. [68Ga]FSC-(RGD)3 a trimeric RGD peptide for imaging αvβ3 integrin expression based on a novel siderophore derived chelating scaffold-synthesis and evaluation. Nucl. Med. Biol. 2015, 42, 115–122. [Google Scholar] [CrossRef]
- Karimi, F.; O’Connor, A.J.; Qiao, G.G.; Heath, D.E. Integrin Clustering Matters: A Review of Biomaterials Functionalized with Multivalent Integrin-Binding Ligands to Improve Cell Adhesion, Migration, Differentiation, Angiogenesis, and Biomedical Device Integration. Adv. Healthc. Mater. 2018, 7, e1701324. [Google Scholar] [CrossRef] [PubMed]
- Pane, K.; Verrillo, M.; Avitabile, A.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Di Maro, A.; Rega, C.; Amoresano, A.; Izzo, V.; et al. Chemical Cleavage of an Asp-Cys Sequence Allows Efficient Production of Recombinant Peptides with an N-Terminal Cysteine Residue. Bioconjug. Chem. 2018, 29, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Litak, P.T.; Kauffman, J.M. Syntheses of reactive fluorescent stains derived from 5(2)-aryl-2(5)-(4-pyridyl)oxazoles and bifunctionally reactive linkers. J. Heterocycl. Chem. 1994, 31, 457–479. [Google Scholar] [CrossRef]
- Dou, Y.; Goodchild, S.J.; Velde, R.V.; Wu, Y.; Fedida, D. The neutral, hydrophobic isoleucine at position I521 in the extracellular S4 domain of hERG contributes to channel gating equilibrium. Am. J. Physiol. Cell Physiol. 2013, 305, 468–478. [Google Scholar] [CrossRef][Green Version]
- Wakabayashi, H.; Fay, P.J. Molecular orientation of Factor VIIIa on the phospholipid membrane surface determined by fluorescence resonance energy transfer. Biochem. J. 2013, 452, 293–301. [Google Scholar] [CrossRef]
- Pane, K.; Durante, L.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Sgambati, V.; Di Maro, A.; Carpentieri, A.; Izzo, V.; Di Donato, A.; et al. Rational design of a carrier protein for the production of recombinant toxic peptides in Escherichia coli. PLoS ONE 2016, 11, e0146552. [Google Scholar] [CrossRef]
- Doose, S.; Neuweiler, H.; Sauer, M. Fluorescence quenching by photoinduced electron transfer: A reporter for conformational dynamics of macromolecules. ChemPhysChem 2009, 10, 1389–1398. [Google Scholar] [CrossRef]
- Chen, H.; Ahsan, S.S.; Santiago-Berrios, M.B.; Abruña, H.D.; Webb, W.W. Mechanisms of quenching of alexa fluorophores by natural amino acids. J. Am. Chem. Soc. 2010, 132, 7244–7245. [Google Scholar] [CrossRef]
- Goldstein, L.; Levin, Y.; Katchalski, E. A Water-insoluble Polyanionic Derivative of Trypsin. II. Effect of the Polyelectrolyte Carrier on the Kinetic Behavior of the Bound Trypsin. Biochemistry 1964, 3, 1913–1919. [Google Scholar] [CrossRef]
- Maurel, P.; Douzou, P. Catalytic implications of electrostatic potentials: The lytic activity of lysozyme as a model. J. Mol. Biol. 1976, 102, 253–264. [Google Scholar] [CrossRef]
- Gaglione, R.; Smaldone, G.; Cesaro, A.; Rumolo, M.; De Luca, M.; Di Girolamo, R.; Petraccone, L.; Del Vecchio, P.; Oliva, R.; Notomista, E.; et al. Impact of a Single Point Mutation on the Antimicrobial and Fibrillogenic Properties of Cryptides from Human Apolipoprotein B. Pharmaceuticals 2021, 14, 631. [Google Scholar] [CrossRef]
- Oliva, R.; Del Vecchio, P.; Grimaldi, A.; Notomista, E.; Cafaro, V.; Pane, K.; Schuabb, V.; Winter, R.; Petraccone, L. Membrane disintegration by the antimicrobial peptide (P)GKY20: Lipid segregation and domain formation. Phys. Chem. Chem. Phys. 2019, 21, 3989–3998. [Google Scholar] [CrossRef]
- Aniansson, E.A.G.; Wall, S.N.; Almgren, M.; Hoffmann, H.; Kielmann, I.; Ulbricht, W.; Zana, R.; Lang, J.; Tondre, C. Theory of the kinetics of micellar equilibria and quantitative interpretation of chemical relaxation studies of micellar solutions of ionic surfactants. J. Phys. Chem. 1976, 80, 905–922. [Google Scholar] [CrossRef]
- Gaglione, R.; Cesaro, A.; Dell’Olmo, E.; Della Ventura, B.; Casillo, A.; Di Girolamo, R.; Velotta, R.; Notomista, E.; Veldhuizen, E.J.A.; Corsaro, M.M.; et al. Effects of human antimicrobial cryptides identified in apolipoprotein B depend on specific features of bacterial strains. Sci. Rep. 2019, 9, 6728. [Google Scholar] [CrossRef]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1513–1522. [Google Scholar] [CrossRef]
- Yu, L.; Tan, M.; Ho, B.; Ding, J.L.; Wohland, T. Determination of critical micelle concentrations and aggregation numbers by fluorescence correlation spectroscopy: Aggregation of a lipopolysaccharide. Anal. Chim. Acta 2006, 556, 216–225. [Google Scholar] [CrossRef]
- Saravanan, R.; Holdbrook, D.A.; Petrlova, J.; Singh, S.; Berglund, N.A.; Choong, Y.K.; Kjellström, S.; Bond, P.J.; Malmsten, M.; Schmidtchen, A. Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides. Nat. Commun. 2018, 9, 2762. [Google Scholar] [CrossRef]
- Claxton, N.S.; Fellers, T.J.; Davidson, M.W. Microscopy, Confocal. In Encyclopedia of Medical Devices and Instrumentation; Webster, J.G., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 449–477. [Google Scholar] [CrossRef]
- Dolman, N.J.; Kilgore, J.A.; Davidson, M.W. A review of reagents for fluorescence microscopy of cellular compartments and structures, part I: BacMam labeling and reagents for vesicular structures. Curr. Protoc. Cytom. 2013, 65, 1–27. [Google Scholar] [CrossRef]
- Nogueira, E.; Cruz, C.F.; Loureiro, A.; Nogueira, P.; Freitas, J.; Moreira, A.; Carmo, A.M.; Gomes, A.C.; Preto, A.; Cavaco-Paulo, A. Assessment of liposome disruption to quantify drug delivery in vitro. Biochim. Biophys. Acta Biomembr. 2016, 1858, 163–167. [Google Scholar] [CrossRef]
- Gargotti, M.; Lopez-Gonzalez, U.; Byrne, H.J.; Casey, A. Comparative studies of cellular viability levels on 2D and 3D in vitro culture matrices. Cytotechnology 2018, 70, 261–273. [Google Scholar] [CrossRef]
- Dinnen, R.D.; Drew, L.; Petrylak, D.P.; Mao, Y.; Cassai, N.; Szmulewicz, J.; Brandt-Rauf, P.; Fine, R.L. Activation of Targeted Necrosis by a p53 Peptide: A Novel Death Pathway That Circumvents Apoptotic Resistance. J. Biol. Chem. 2007, 282, 26675–26686. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Mana, G.; Valdembri, D.; Serini, G. Conformationally active integrin endocytosis and traffic: Why, where, when and how? Biochem. Soc. Trans. 2020, 48, 83–93. [Google Scholar] [CrossRef]
- Buckley, C.D.; Pilling, D.; Henriquez, N.V.; Parsonage, G.; Threlfall, K.; Scheel-Toellner, D.; Simmons, D.L.; Akbar, A.N.; Lord, J.M.; Salmon, M. RGD peptides induce apoptosis by direct caspase-3 activation. Nature 1999, 397, 534–539. [Google Scholar] [CrossRef]
- Webb, J.N.; Koufos, E.; Brown, A.C. Inhibition of Bacterial Toxin Activity by the Nuclear Stain, DRAQ5TM. J. Membr. Biol. 2016, 249, 503–511. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Elliott, T.; Holt, M.; Muir, T.W.; Chin, J.W. Genetically encoded 1,2-aminothiols facilitate rapid and site-specific protein labeling via a bio-orthogonal cyanobenzothiazole condensation. J. Am. Chem. Soc. 2011, 133, 11418–11421. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, X.; Mei, B.; Zhang, D.; Tang, A.; An, L.; He, X.; Jiang, J.; Liang, G. Labeling thiols on proteins, living cells, and tissues with enhanced emission induced by FRET. Sci. Rep. 2013, 3, 3523. [Google Scholar] [CrossRef]
- Koniev, O.; Wagner, A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. [Google Scholar] [CrossRef]
- Takakura, H. Molecular Design of d-Luciferin-Based Bioluminescence and 1,2-Dioxetane-Based Chemiluminescence Substrates for Altered Output Wavelength and Detecting Various Molecules. Molecules 2021, 26, 1618. [Google Scholar] [CrossRef]
- Sharma, D.K.; Adams, S.T.; Liebmann, K.L.; Miller, S.C. Rapid Access to a Broad Range of 6′-Substituted Firefly Luciferin Analogues Reveals Surprising Emitters and Inhibitors. Org. Lett. 2017, 19, 5836–5839. [Google Scholar] [CrossRef]
- Mofford, D.M.; Reddy, G.R.; Miller, S.C. Aminoluciferins extend firefly luciferase bioluminescence into the near-infrared and can be preferred substrates over d-luciferin. J. Am. Chem. Soc. 2014, 136, 13277–13282. [Google Scholar] [CrossRef] [PubMed]
- Takakura, H.; Kojima, R.; Ozawa, T.; Nagano, T.; Urano, Y. Development of 5′- and 7′-Substituted Luciferin Analogues as Acid-Tolerant Substrates of Firefly Luciferase. ChemBioChem 2012, 13, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Fery-Forgues, S.; Lavabre, D. Are Fluorescence Quantum Yields So Tricky to Measure? A Demonstration Using Familiar Stationery Products. J. Chem. Educ. 1999, 76, 1260. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
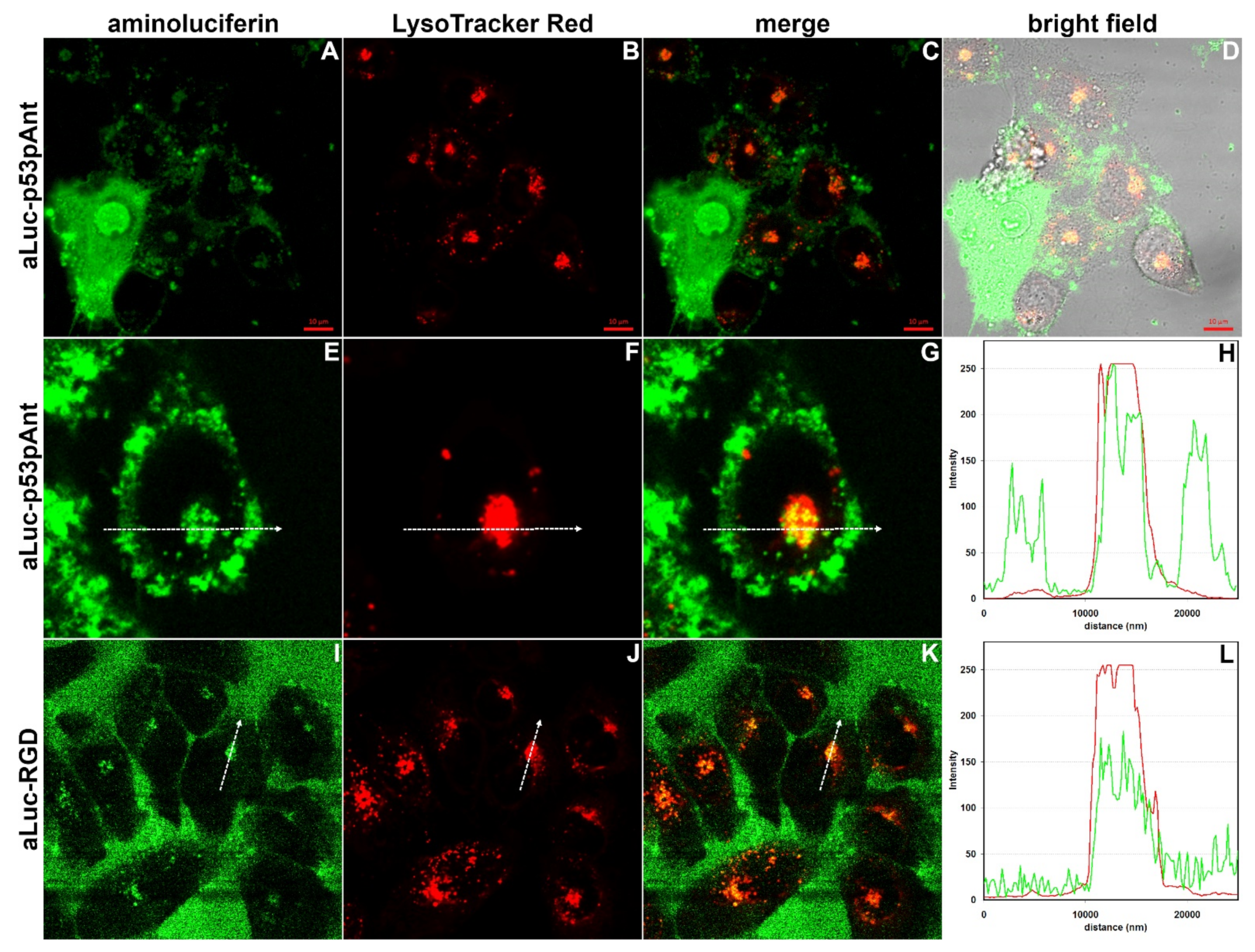{kind=link}
| Peptide | λmax (nm) a | |||||||
|---|---|---|---|---|---|---|---|---|
| NaP pH 7.4 | MeOH 50% | IPA 50% | SDS (25 mM) | POPC +POPG | POPC | LPS (200 µg/mL) | E. coli Cells | |
| Luc-GKY20 | 538 | 533 (5) | 531 (7) | 526 (12) | 520 (19) | 520 (19) | 516 (23) | 522 (16) |
| Luc-ApoBL | 539 | 533 (6) | 533 (6) | 526 (13) | 522 (17) | 539 (0) | 508 (31) | 534 (5) |
| Luc-p53pAnt | 539 | 533 (6) | 533 (6) | 526 (13) | 522 (17) | 539 (0) | nd b | nd |
| Luc-RGD | 539 | 533 (6) | 533 (6) | 533 (6) | 539 (0) | 539 (0) | nd | nd |
| aLuc-GKY20 | 525 | 515 (10) | 510 (15) | 506 (19) | 499 (26) | 499 (26) | 503 (22) | 509 (16) |
| aLuc-ApoBL | 527 | 516 (11) | 512 (15) | 505 (22) | 497 (27) | 527 (0) | 486 (41) | 518 (9) |
| aLuc-p53pAnt | 526 | 516 (10) | 515 (11) | 504 (22) | 499 (25) | 526 (0) | nd | nd |
| aLuc-RGD | 526 | 517 (9) | 513 (13) | 510 (16) | 526 (0) | 526 (0) | nd | nd |
| mLuc-GKY20 | 439 | 429 (10) | 424 (15) | 422 (17) | nd | nd | nd | nd |
| PyMPO-(C)GKY20 | 563 | 563 (0) | 558 (5) | 556 (7) | 535 (28) | 541 (22) | nd | nd |
| Peptide | QY a (Variation Relative to NaP pH 7.4) | ||
|---|---|---|---|
| NaP pH 7.4 | IPA 50% b | NaAc c pH 5.0 | |
| PyMPO-GKY20 | 0.080 | 0.373 (4.66) | ndd |
| mLuc-GKY20 | 0.008 | 0.021 (2.63) | nd |
| Luc-GKY20 | 0.111 | 0.639 (5.76) | 0.269 (2.42) |
| aLuc-GKY20 | 0.202 | 0.969 (4.80) | nd |
| Luc-ApoBL | 0.365 | 0.389 (1.07) | 0.450 (1.23) |
| aLuc-ApoBL | 0.574 | 0.757 (1.32) | nd |
| Luc-p53pAnt | 0.261 | 0.184 (0.70) | 0.418 (1.60) |
| aLuc-p53pAnt | 0.432 | 0.274 (0.63) | nd |
| Luc-RGD | 0.519 | 0.443 (0.85) | 0.510 (0.98) |
| aLuc-RGD | 0.937 | 1.040 (1.11) | nd |
| Peptide | Net Charge of the Peptidyl Moiety a | pKa |
|---|---|---|
| Free Luc | not applicable | 8.70 b |
| Luc-RGD | −1 | 8.40 ± 0.03 |
| Luc-GKY20 | +4 | 8.13 ± 0.04 |
| Luc-ApoBL | +6 | 8.11 ± 0.04 |
| Luc-p53pAnt | +11 | 7.97 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siepi, M.; Oliva, R.; Masino, A.; Gaglione, R.; Arciello, A.; Russo, R.; Di Maro, A.; Zanfardino, A.; Varcamonti, M.; Petraccone, L.; et al. Environment-Sensitive Fluorescent Labelling of Peptides by Luciferin Analogues. Int. J. Mol. Sci. 2021, 22, 13312. https://doi.org/10.3390/ijms222413312
Siepi M, Oliva R, Masino A, Gaglione R, Arciello A, Russo R, Di Maro A, Zanfardino A, Varcamonti M, Petraccone L, et al. Environment-Sensitive Fluorescent Labelling of Peptides by Luciferin Analogues. International Journal of Molecular Sciences. 2021; 22(24):13312. https://doi.org/10.3390/ijms222413312
Chicago/Turabian StyleSiepi, Marialuisa, Rosario Oliva, Antonio Masino, Rosa Gaglione, Angela Arciello, Rosita Russo, Antimo Di Maro, Anna Zanfardino, Mario Varcamonti, Luigi Petraccone, and et al. 2021. "Environment-Sensitive Fluorescent Labelling of Peptides by Luciferin Analogues" International Journal of Molecular Sciences 22, no. 24: 13312. https://doi.org/10.3390/ijms222413312
APA StyleSiepi, M., Oliva, R., Masino, A., Gaglione, R., Arciello, A., Russo, R., Di Maro, A., Zanfardino, A., Varcamonti, M., Petraccone, L., Del Vecchio, P., Merola, M., Pizzo, E., Notomista, E., & Cafaro, V. (2021). Environment-Sensitive Fluorescent Labelling of Peptides by Luciferin Analogues. International Journal of Molecular Sciences, 22(24), 13312. https://doi.org/10.3390/ijms222413312