Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Biological Basis of Glioblastoma
2.1. Epidemiology and Classification
2.2. Pathogenesis: Molecular Features and Genomic Alteration
2.3. Heterogeneity
2.4. Tumor Microenvironment
2.5. Crosstalk between Tumor Cells and Their Microenvironment
2.6. Diagnosis and Current Therapy Approach
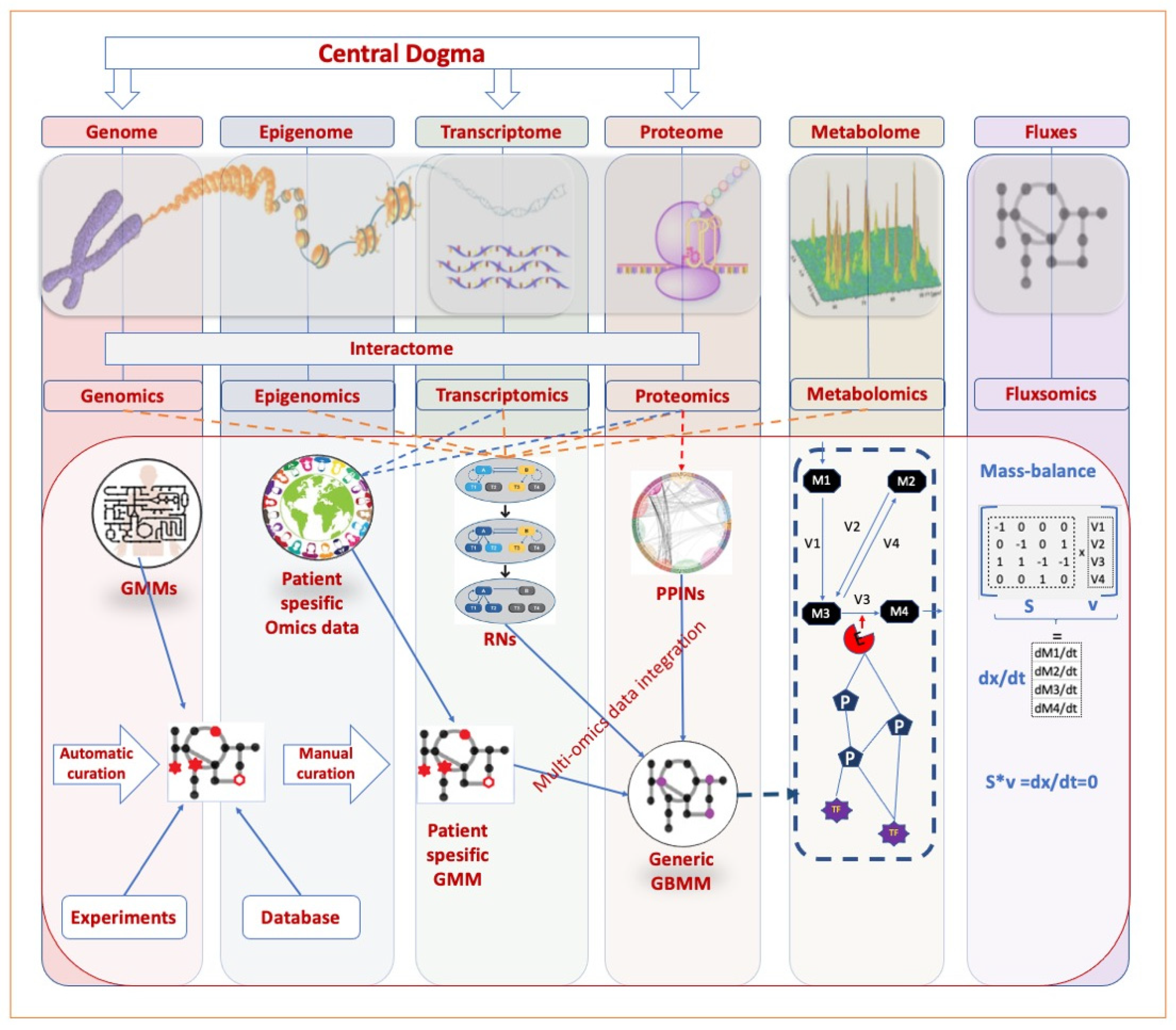
3. Systems Biology Approaches to GBM
3.1. Omics Data
3.1.1. Transcriptomics
- Microarray-based transcriptomics of GBM
- b.
- Next-Generation Sequencing based transcriptomics
3.1.2. Proteomics Data
3.2. Network Analysis of GBM
3.3. Genome-Scale Metabolic Network Modeling
3.4. GBM Related Systems Biology Studies
3.5. Examples of Systems Biology Studies in Other Cancers
3.5.1. Kidney Cancer
3.5.2. HCC
3.5.3. Drug Repositioning Applications in GBM
4. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Fletcher, M.; Gu, Z.; Wang, Q.; Costa, B.; Bertoni, A.; Man, K.H.; Schlotter, M.; Felsberg, J.; Mangei, J.; et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat. Commun. 2020, 11, 6434. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.; Walter, F.M.; Rubin, G.; Neal, R.D. Improving early diagnosis of symptomatic cancer. Nat. Rev. Clin. Oncol. 2016, 13, 740–749. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Exon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schurer, S.C.; Ayad, N.G. Drug Repositioning in Glioblastoma: A Pathway Perspective. Front. Pharm. 2018, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turanli, B.; Altay, O.; Borén, J.; Turkez, H.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Systems biology based drug repositioning for development of cancer therapy. Semin. Cancer Biol. 2021, 68, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab. Investig. 2021, 1–8. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Eder, K.; Kalman, B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol. Oncol. Res. 2014, 20, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Prolo, L.M.; Li, A.; Owen, S.F.; Parker, J.J.; Foshay, K.; Nitta, R.T.; Morgens, D.W.; Bolin, S.; Wilson, C.M.; Luo, E.J.; et al. Targeted genomic CRISPR-Cas9 screen identifies MAP4K4 as essential for glioblastoma invasion. Sci. Rep. 2019, 9, 14020. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Khong, P.; Parkinson, J.F.; Howell, V.M.; Wheeler, H.R. Molecular heterogeneity in glioblastoma: Potential clinical implications. Front. Oncol. 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.M.; Huang, W.; Park, R.; Park, P.J.; Johnson, M.D. A developmental taxonomy of glioblastoma defined and maintained by MicroRNAs. Cancer Res. 2011, 71, 3387–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Exon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Matias, D.; Balca-Silva, J.; da Graca, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Cancer-neuronal crosstalk and the startups working to silence it. Nat. Biotechnol. 2020, 38, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. BioMed Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Oliva, R.; Dominguez-Garcia, S.; Carrascal, L.; Abalos-Martinez, J.; Pardillo-Diaz, R.; Verastegui, C.; Castro, C.; Nunez-Abades, P.; Geribaldi-Doldan, N. Evolution of Experimental Models in the Study of Glioblastoma: Toward Finding Efficient Treatments. Front. Oncol. 2020, 10, 614295. [Google Scholar] [CrossRef]
- Robertson, F.L.; Marques-Torrejon, M.A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis. Model. Mech. 2019, 12, dmm040386. [Google Scholar] [CrossRef] [Green Version]
- Altay, O.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Systems biology perspective for studying the gut microbiota in human physiology and liver diseases. EBioMedicine 2019, 49, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, S.; Hartmann, N.; Benfeitas, R.; Zhang, C.; Arif, M.; Turkez, H.; Uhlén, M.; Englert, C.; Knight, R.; Mardinoglu, A. Systems Analysis Reveals Ageing-Related Perturbations in Retinoids and Sex Hormones in Alzheimer’s and Parkinson’s Diseases. Biomedicines 2021, 9, 1310. [Google Scholar] [CrossRef]
- Grizzle, W.E.; Bell, W.C.; Sexton, K.C. Issues in collecting, processing and storing human tissues and associated information to support biomedical research. Cancer Biomark. 2010, 9, 531–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst Biol. 2014, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast reconstruction of compact context-specific metabolic network models. PLoS Comput. Biol. 2014, 10, e1003424. [Google Scholar] [CrossRef]
- Kim, M.K.; Lane, A.; Kelley, J.J.; Lun, D.S. E-Flux2 and SPOT: Validated Methods for Inferring Intracellular Metabolic Flux Distributions from Transcriptomic Data. PLoS ONE 2016, 11, e0157101. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2010, 27, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.J.; Ebrahim, A.; Metz, T.O.; Adkins, J.N.; Palsson, B.O.; Hyduke, D.R. GIM3E: Condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 2013, 29, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, P.; Mardinoglu, A.; Nielsen, J. Cancer Metabolism: A Modeling Perspective. Front. Physiol. 2015, 6, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, I.; Uhlen, M.; Zhang, C.; Mardinoglu, A. Genome-Scale Metabolic Modeling of Glioblastoma Reveals Promising Targets for Drug Development. Front. Genet. 2020, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Zhang, C.; Kilicarslan, M.; Piening, B.D.; Bjornson, E.; Hallström, B.M.; Groen, A.K.; Ferrannini, E.; Laakso, M.; Snyder, M.; et al. Integrated Network Analysis Reveals an Association between Plasma Mannose Levels and Insulin Resistance. Cell Metab. 2016, 24, 172–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, A.; Keener, C.; Boynton, A.L.; Warrick, J.; Murphy, G.P. CXCR-4, a chemokine receptor, is overexpressed in and required for proliferation of glioblastoma tumor cells. J. Surg. Oncol. 1998, 69, 99–104. [Google Scholar] [CrossRef]
- Sallinen, S.L.; Sallinen, P.K.; Haapasalo, H.K.; Helin, H.J.; Helen, P.T.; Schraml, P.; Kallioniemi, O.P.; Kononen, J. Identification of differentially expressed genes in human gliomas by DNA microarray and tissue chip techniques. Cancer Res. 2000, 60, 6617–6622. [Google Scholar] [PubMed]
- Ma, D.; Nutt, C.L.; Shanehsaz, P.; Peng, X.; Louis, D.N.; Kaetzel, D.M. Autocrine platelet-derived growth factor-dependent gene expression in glioblastoma cells is mediated largely by activation of the transcription factor sterol regulatory element binding protein and is associated with altered genotype and patient survival in human brain tumors. Cancer Res. 2005, 65, 5523–5534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.Y.; Wei, K.C.; Huang, A.C.; Wang, K.; Huang, C.Y.; Yi, D.; Tang, C.Y.; Galas, D.J.; Hood, L.E. RNASEQR--a streamlined and accurate RNA-seq sequence analysis program. Nucleic Acids Res. 2012, 40, e42. [Google Scholar] [CrossRef]
- Gargiulo, G.; Cesaroni, M.; Serresi, M.; de Vries, N.; Hulsman, D.; Bruggeman, S.W.; Lancini, C.; van Lohuizen, M. In vivo RNAi screen for BMI1 targets identifies TGF-beta/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell 2013, 23, 660–676. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Dix, M.M.; Cravatt, B.F. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat. Rev. Cancer 2010, 10, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Deighton, R.F.; Le Bihan, T.; Martin, S.F.; Gerth, A.M.J.; McCulloch, M.; Edgar, J.M.; Kerr, L.E.; Whittle, I.R.; McCulloch, J. Interactions among mitochondrial proteins altered in glioblastoma. J. Neurooncol. 2014, 118, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Mahalingam, K. Comprehensive analysis of Reverse Phase Protein Array data reveals characteristic unique proteomic signatures for glioblastoma subtypes. Gene 2019, 685, 85–95. [Google Scholar] [CrossRef]
- Wang, X.; Bustos, M.A.; Zhang, X.; Ramos, R.I.; Tan, C.; Iida, Y.; Chang, S.C.; Salomon, M.P.; Tran, K.; Gentry, R.; et al. Downregulation of the Ubiquitin-E3 Ligase RNF123 Promotes Upregulation of the NF-kappaB1 Target SerpinE1 in Aggressive Glioblastoma Tumors. Cancers 2020, 12, 81. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Nielsen, J. Systems medicine and metabolic modelling. J. Intern. Med. 2012, 271, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, E.; Cakir, T. Reconstructed Metabolic Network Models Predict Flux-Level Metabolic Reprogramming in Glioblastoma. Front. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, P. GEPASI: A software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 1993, 9, 563–571. [Google Scholar] [CrossRef]
- Sauro, H.M. SCAMP: A general-purpose simulator and metabolic control analysis program. Comput. Appl. Biosci. 1993, 9, 441–450. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Yurkovich, J.T.; Paglia, G.; Rolfsson, O.; Sigurjonsson, O.E.; Palsson, B.O. Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 2017, 7, 46249. [Google Scholar] [CrossRef] [Green Version]
- Jamialahmadi, O.; Hashemi-Najafabadi, S.; Motamedian, E.; Romeo, S.; Bagheri, F. A benchmark-driven approach to reconstruct metabolic networks for studying cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006936. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Sertbas, M.; Ulgen, K.; Cakir, T. Systematic analysis of transcription-level effects of neurodegenerative diseases on human brain metabolism by a newly reconstructed brain-specific metabolic network. FEBS Open Biol. 2014, 4, 542–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Kocabas, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.O. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Drager, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Lewis, N.E.; Schramm, G.; Bordbar, A.; Schellenberger, J.; Andersen, M.P.; Cheng, J.K.; Patel, N.; Yee, A.; Lewis, R.A.; Eils, R.; et al. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010, 28, 1279–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat. Commun. 2017, 8, 14250. [Google Scholar] [CrossRef]
- Skiriute, D.; Vaitkiene, P.; Saferis, V.; Asmoniene, V.; Skauminas, K.; Deltuva, V.P.; Tamasauskas, A. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Tachibana, H.; Ishiyama, Y.; Yoshino, M.; Yamashita, K.; Toki, D.; Kondo, T. Efficacy of Cabozantinib in Metastatic Papillary Renal Cell Carcinoma Following Ineffective Treatment With Initial Therapy of Nivolumab and Ipilimumab. In Vivo 2021, 35, 1743–1747. [Google Scholar] [CrossRef]
- Brannon, A.R.; Reddy, A.; Seiler, M.; Arreola, A.; Moore, D.T.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Liu, H.; Nathanson, K.L.; et al. Molecular Stratification of Clear Cell Renal Cell Carcinoma by Consensus Clustering Reveals Distinct Subtypes and Survival Patterns. Genes Cancer 2010, 1, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Kim, W.; Juszczak, K.; Arif, M.; Sato, Y.; Kume, H.; Ogawa, S.; Turkez, H.; Boren, J.; Nielsen, J.; et al. Stratification of patients with clear cell renal cell carcinoma to facilitate drug repositioning. iScience 2021, 24, 102722. [Google Scholar] [CrossRef] [PubMed]
- Koudijs, K.K.M.; van Scheltinga, A.G.T.T.; Bohringer, S.; Schimmel, K.J.M.; Guchelaar, H.J. Personalised drug repositioning for Clear Cell Renal Cell Carcinoma using gene expression. Sci. Rep. 2018, 8, 5250. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Turanli, B.; Juszczak, K.; Kim, W.; Arif, M.; Sato, Y.; Ogawa, S.; Turkez, H.; Nielsen, J.; Boren, J.; et al. Classification of clear cell renal cell carcinoma based on PKM alternative splicing. Heliyon 2020, 6, e03440. [Google Scholar] [CrossRef]
- Zerbini, L.F.; Bhasin, M.K.; de Vasconcellos, J.F.; Paccez, J.D.; Gu, X.; Kung, A.L.; Libermann, T.A. Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol. Cancer Ther. 2014, 13, 1929–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Turanli, B.; Grotli, M.; Boren, J.; Nielsen, J.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Drug Repositioning for Effective Prostate Cancer Treatment. Front. Physiol. 2018, 9, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turanli, B.; Zhang, C.; Kim, W.; Benfeitas, R.; Uhlen, M.; Arga, K.Y.; Mardinoglu, A. Discovery of therapeutic agents for prostate cancer using genome-scale metabolic modeling and drug repositioning. EBioMedicine 2019, 42, 386–396. [Google Scholar] [CrossRef] [Green Version]
- Altay, O.; Zhang, C.; Turkez, H.; Nielsen, J.; Uhlén, M.; Mardinoglu, A. Revealing the Metabolic Alterations during Biofilm Development of Burkholderia cenocepacia Based on Genome-Scale Metabolic Modeling. Metabolites 2021, 11, 221. [Google Scholar] [CrossRef]
- Turanli, B.; Karagoz, K.; Bidkhori, G.; Sinha, R.; Gatza, M.L.; Uhlen, M.; Mardinoglu, A.; Arga, K.Y. Multi-Omic Data Interpretation to Repurpose Subtype Specific Drug Candidates for Breast Cancer. Front. Genet. 2019, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, C.S.; Voight, B.F. Pathway and network-based strategies to translate genetic discoveries into effective therapies. Hum. Mol. Genet. 2016, 25, R94–R98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanseau, P.; Agarwal, P.; Barnes, M.R.; Pastinen, T.; Richards, J.B.; Cardon, L.R.; Mooser, V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012, 30, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef]
- Fahey, E.J.; Pomeroy, E.; Rowan, F.E. Acute trunnion failure of a TMZF alloy stem with large diameter femoral heads. J. Orthop. 2020, 20, 17–20. [Google Scholar] [CrossRef]
- Benavides-Serrato, A.; Saunders, J.T.; Holmes, B.; Nishimura, R.N.; Lichtenstein, A.; Gera, J. Repurposing Potential of Riluzole as an ITAF Inhibitor in mTOR Therapy Resistant Glioblastoma. Int. J. Mol. Sci. 2020, 21, 344. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Resources | Omics Layer | Notes | Reference |
|---|---|---|---|
| Projects | |||
| TCGA | Genomics, Proteomics, Transcriptomics, Epigenomics | http://cancergenome.nih.gov/, accessed on 29 October 2021 | |
| CGP | Genomics, Proteomics, Transcriptomics, Epigenomics | https://www.sanger.ac.uk/, accessed on 25 October 2021 | |
| ICGC | Genomics, Transcriptomics, Epigenomics | https://daco.icgc.org/, accessed on 23 October 2021 | |
| CPTAC | Proteomics, Genomics | https://proteomics.cancer.gov/, accessed on 19 October 2021 | |
| Databases | |||
| GEO | Genomics, Transcriptomic | https://www.ncbi.nlm.nih.gov/geo/, accessed on 19 October 2021 | |
| Expression Atlas | Genomics, Proteomics, Transcriptomics, Epigenomics, Interactomics | https://www.ebi.ac.uk/gxa/home, accessed on 29 October 2021 | |
| ArrayExpress | Genomics, Proteomics, Transcriptomics, Epigenomics, Interactomics | https://www.ebi.ac.uk/arrayexpress/, accessed on 23 October 2021 | |
| Human Protein Atlas | Proteomics, Transcriptomics | https://www.proteinatlas.org/, accessed on 25 October 2021 | |
| DDBJ | Genomics, Transcriptomics | https://www.ddbj.nig.ac.jp/, accessed on 24 October 2021 | |
| ENCODE | Genomics, Transcriptomics, Epigenomics | https://www.encodeproject.org/, accessed on 19 October 2021 | |
| StarBase | Interactomics | Pathway browser, Analysis tools | http://starbase.sysu.edu.cn/, accessed on 29 October 2021 |
| BioGrid | Interactomics | Biological interaction, PPI | https://thebiogrid.org/, accessed on 22 October 2021 |
| Reactome | Genomics, Proteomics, Transcriptomics, Interactomics | Reactions, Pathway browser, Analysis tools, Visualization | https://reactome.org/, accessed on 29 October 2021 |
| KEGG | Proteomics, Transcriptomics, Proteomics, Interactomics | Reactions, Pathway browser, Analysis tools, Visualization | https://www.genome.jp/kegg/, accessed on 29 October 2021 |
| STRING | Interactomics | Pathway browser, Analysis tools, Visualization | https://string-db.org/, accessed on 15 October 2021 |
| HMDB | metabolomics | Pathway browser, Analysis tools | https://hmdb.ca/, accessed on 13 October 2021 |
| GeneBank | Genomics | Analysis tools | https://www.ncbi.nlm.nih.gov/genbank/, accessed on 21 October 2021 |
| Ensembl | Genomics | Genome browser, comparative genomics, Analysis tools | https://www.ensembl.org/, accessed on 21 October 2021 |
| PRIDE | Proteomics | Analysis tools | https://www.ebi.ac.uk/pride/, accessed on 24 October 2021 |
| Lipid Maps | Lipidomics | Analysis tools, Structure drawing | https://www.lipidmaps.org/, accessed on 29 October 2021 |
| UniProt | Proteomics | Analysis tools | https://www.uniprot.org/, accessed on 29 October 2021 |
| ChEBI | Metabolomics | Small chemical compounds | https://www.ebi.ac.uk/chebi/, accessed on 29 October 2021 |
| MetaboLights | Metabolomics | Metabolomics repository | https://www.ebi.ac.uk/metabolights/, accessed on 29 October 2021 |
| JASPAR | Interactomics | TF binding, Analysis tools | http://jaspar.genereg.net/, accessed on 25 October 2021 |
| geneXplain | Interactomics | Analysis tools, TF binding | https://genexplain.com/, accessed on 22 October 2021 |
| HPRD | Proteomics, Interactomics | Pathway browser, Analysis tools, PPI | http://www.hprd.org/, accessed on 18 October 2021 |
| miRTarBase | Interactomics | miRNA-target interactions | http://miRTarBase.cuhk.edu.cn/, accessed on 12 October 2021 |
| GWAS Catalog | Genomics | Genetic variant | https://www.ebi.ac.uk/gwas/, accessed on 19 October 2021 |
| dbGAP | Genomics, Epigenomics | Genotypes and Phenotypes, Analysis tools | https://www.ncbi.nlm.nih.gov/gap/, accessed on 19 October 2021 |
| dbSNP | Genomics | SNP genotyping | https://www.ncbi.nlm.nih.gov/snp/, accessed on 19 October 2021 |
| Tools | |||
| 3Omics | Transcriptomics, Proteomics, Metabolomics | Pathway enrichment, correlation and co-expression network, ID conversion | https://3omics.cmdm.tw/, accessed on 29 October 2021 |
| BioCyc and MetaCyc | Genomics, Proteomics, Metabolomics | Pathway, Enzymes, Reactions, Analysis tools | https://biocyc.org/, accessed on 23 October 2021 |
| Cell Illustrator 5.0 | Genomics, Transcriptomics, Proteomics | Visualize biological pathways | http://www.cellillustrator.com/home, accessed on 29 October 2021 |
| CellML | Genomics, Transcriptomics, Proteomics | Mathematical modeling, XML markup language | https://www.cellml.org/, accessed on 21 October 2021 |
| COBRA | Genomics, Transcriptomics, Proteomics | Constraint-based modeling, MATLAB | https://opencobra.github.io/cobratoolbox/stable/, accessed on 22 October 2021 |
| RAVEN 2.0 | Genomics, Proteomics | Genome-scale metabolic modeling, MATLAB | https://github.com/SysBioChalmers/RAVEN, accessed on 22 October 2021 |
| Cytoscape | Genomics, Transcriptomics, Proteomics, Fluxomics | Visualizing and integrating pathways | https://cytoscape.org/, accessed on 14 October 2021 |
| E-Cell | Genomics, Transcriptomics, Proteomics | Modeling, simulation, and analysis | https://www.e-cell.org/, accessed on 12 October 2021 |
| Escher | Genomics, Proteomics, Metabolomics | Visualization of metabolic pathways | https://escher.github.io/#/, accessed on 19 October 2021 |
| Gaggle | Genomics, Transcriptomics, Proteomics, Fluxomics | Integration of diverse database | https://isbscience.org/, accessed on 19 October 2021 |
| IMPaLA | Transcriptomics, Proteomics, Metabolomics | Pathway analysis | http://impala.molgen.mpg.de/, accessed on 19 October 2021 |
| Ingenuity Pathway Analysis | Transcriptomics, Proteomics, Metabolomics | Pathway analysis, commercial | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/, accessed on 25 October 2021 |
| MarVis-Pathway | Transcriptomics, Metabolomics | Pathway browser, Visualization | http://marvis.gobics.de/, accessed on 25 October 2021 |
| MassTrix | Metabolomics, Proteomics | Mapping, Analysis | http://masstrix.org/, accessed on 23 October 2021 |
| MetaboAnalyst | Genomics, Transcriptomics, Proteomics, Metabolomics | Integrative Analysis | http://www.metaboanalyst.ca/, accessed on 29 October 2021 |
| MetaboLights | Metabolomics | Database | http://www.ebi.ac.uk/metabolights/, accessed on 29 October 2021 |
| MetScape 3 | Transcriptomics, Metabolomics | Visualization, interpretation | http://metscape.ncibi.org/, accessed on 29 October 2021 |
| mixOmics | Transcriptomics, Proteomics, Metabolomics | Integration and exploration of datasets | http://mixomics.org/, accessed on 29 October 2021 |
| OmicsPLS | Transcriptomics, Proteomics, Metabolomics | Data integration, R | https://github.com/cran/OmicsPLS, accessed on 29 October 2021 |
| Omickriging | Transcriptomics, Proteomics, Metabolomics, Fluxomics | Omics integration tools, R | https://cran.r-project.org/web/packages/OmicKriging/index.html, accessed on 21 October 2021 |
| Omix visualization tool | Transcriptomics, Proteomics, Metabolomics, Fluxomics | Visualization and modeling, commercial | https://www.omix-visualization.com/#sthash.ScUNDhbD.dpbs, accessed on 21 October 2021 |
| PaintOmics 3 | Transcriptomics, Metabolomics | Integrative visualization | http://www.paintomics.org/, accessed on 29 October 2021 |
| PathVisio 3 | Transcriptomics, Proteomics, Metabolomics | Pathway creation and curation | https://pathvisio.github.io/, accessed on 21 October 2021 |
| SimCell | Genomics, Proteomics, Transcriptomics, Metabolomics | Cell simulation | http://wishart.biology.ualberta.ca/SimCell/, accessed on 29 October 2021 |
| VANTED | Transcriptomics, Proteomics, Metabolomics | Mapping, Processing, Analysis, Visualization | https://www.cls.uni-konstanz.de/software/vanted/, accessed on 24 October 2021 |
| Omics data integration methods for GEMs | |||
| tINIT | Transcriptomics, Proteomics | Task-driven model reconstruction algorithm | [34] |
| FASTCORE | Transcriptomics | Context specific metabolic modeling | [35] |
| E-Flux2 | Transcriptomics | Infers fluxes from transcriptomic data | [36] |
| SPOT | Transcriptomics | Correlation between fluxes and enzymatic transcript | [36] |
| PROM | Transcriptomics | The probability of a gene being on-off in the inactivation of a TF | [37] |
| MADE | Transcriptomics, Proteomics | The algorithm uses DEG genes or proteins to generate a GEM | [38] |
| GIM3E | Transcriptomics, Proteomics, Metabolomics | An algorithm creates a condition-specific metabolic network based on the objective function, transcriptome, and metabolome | [39] |
| Current Generic GEMs | Reaction Number | Metabolite Number | Gene Number | References |
|---|---|---|---|---|
| HMR1 | 8100 | 6000 | 3668 | [57] |
| HMR2 | 8181 | 6006 | 3765 | [62] |
| Human 1 | 13,082 | 8378 | 3625 | [63] |
| Recon 1 | 3744 | 2766 | 1905 | [64] |
| Recon 2.2 | 7440 | 5063 | 2140 | [65] |
| Recon 3D | 13,543 | 4140 | 3288 | [66] |
| iNL403 | 1070 | 987 | 403 | [67] |
| iMS570 | 630 | 524 | 570 | [61] |
| iHsa | 8264 | 5620 | 2315 | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaynar, A.; Altay, O.; Li, X.; Zhang, C.; Turkez, H.; Uhlén, M.; Shoaie, S.; Mardinoglu, A. Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 13213. https://doi.org/10.3390/ijms222413213
Kaynar A, Altay O, Li X, Zhang C, Turkez H, Uhlén M, Shoaie S, Mardinoglu A. Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme. International Journal of Molecular Sciences. 2021; 22(24):13213. https://doi.org/10.3390/ijms222413213
Chicago/Turabian StyleKaynar, Ali, Ozlem Altay, Xiangyu Li, Cheng Zhang, Hasan Turkez, Mathias Uhlén, Saeed Shoaie, and Adil Mardinoglu. 2021. "Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme" International Journal of Molecular Sciences 22, no. 24: 13213. https://doi.org/10.3390/ijms222413213
APA StyleKaynar, A., Altay, O., Li, X., Zhang, C., Turkez, H., Uhlén, M., Shoaie, S., & Mardinoglu, A. (2021). Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme. International Journal of Molecular Sciences, 22(24), 13213. https://doi.org/10.3390/ijms222413213