
Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells




Abstract
:1. Introduction
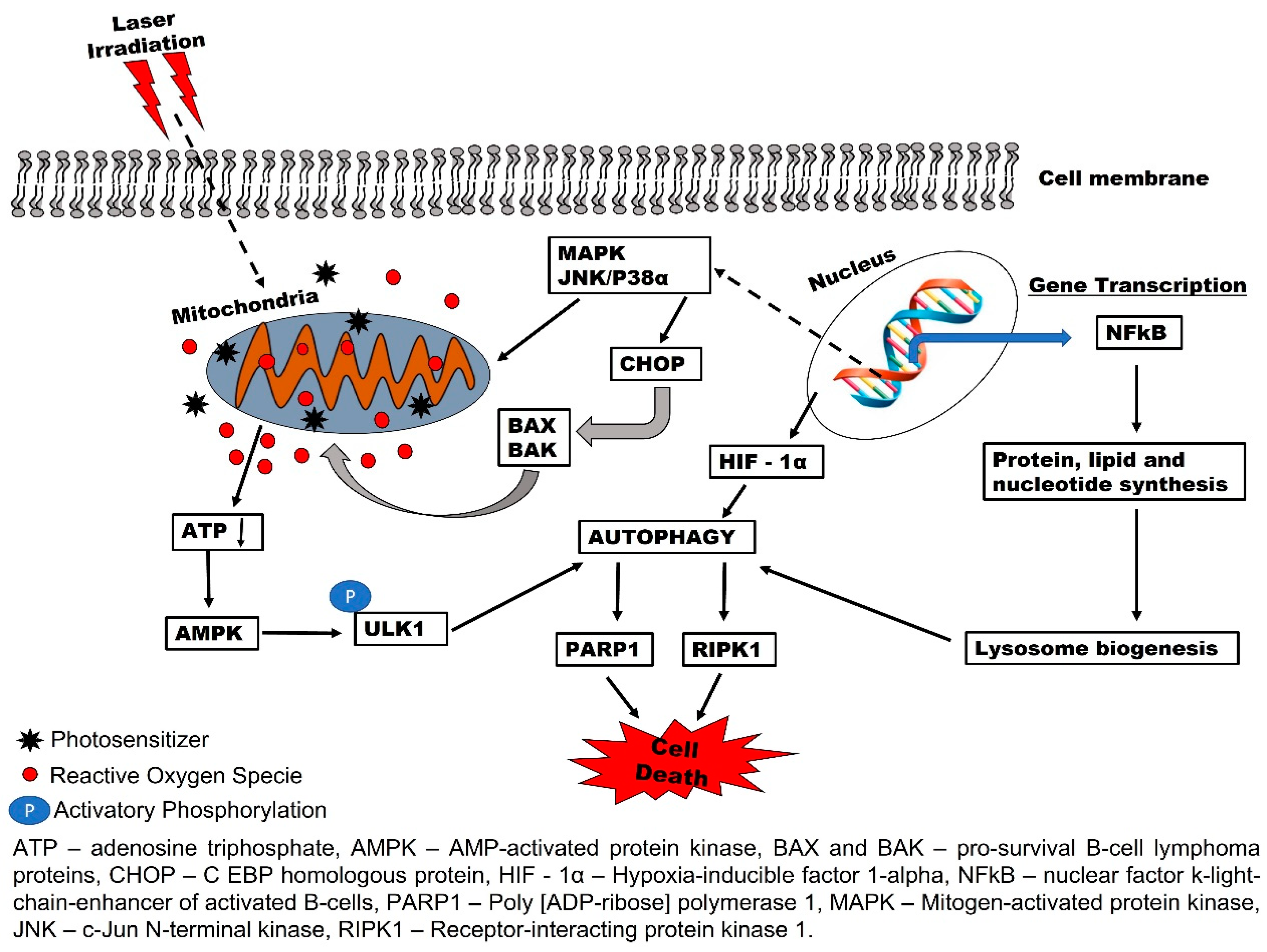
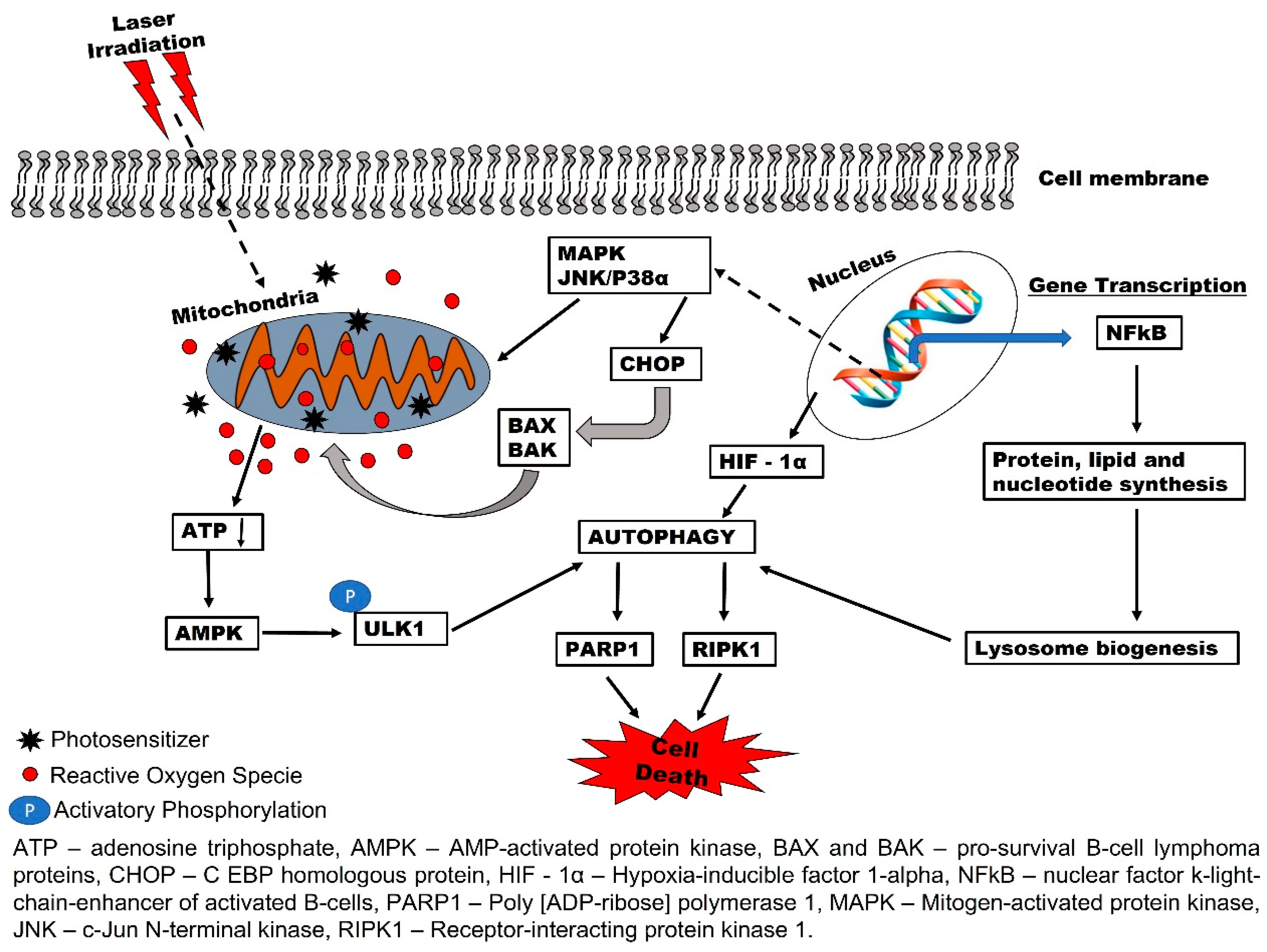
2. Biochemical Effects of PDT and Reactive Oxygen Species (ROS) Production
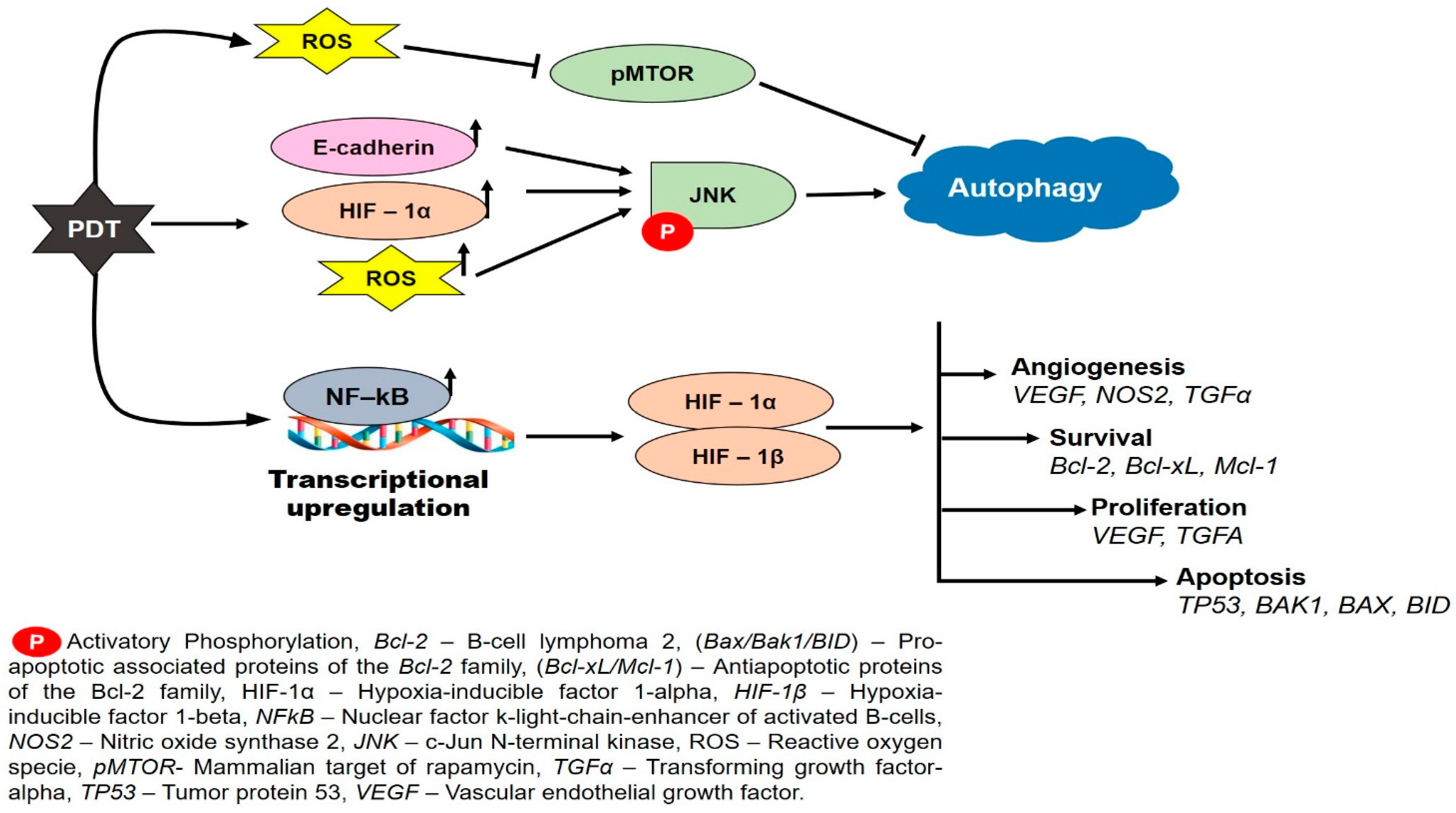
3. Autophagy-Mediated PDT Resistance in Tumor Cells
4. Involvement of Pro-Survival Apoptotic Proteins in Photodynamic Therapy Resistance
5. Hypoxia-Induced Resistance to Photodynamic Therapy
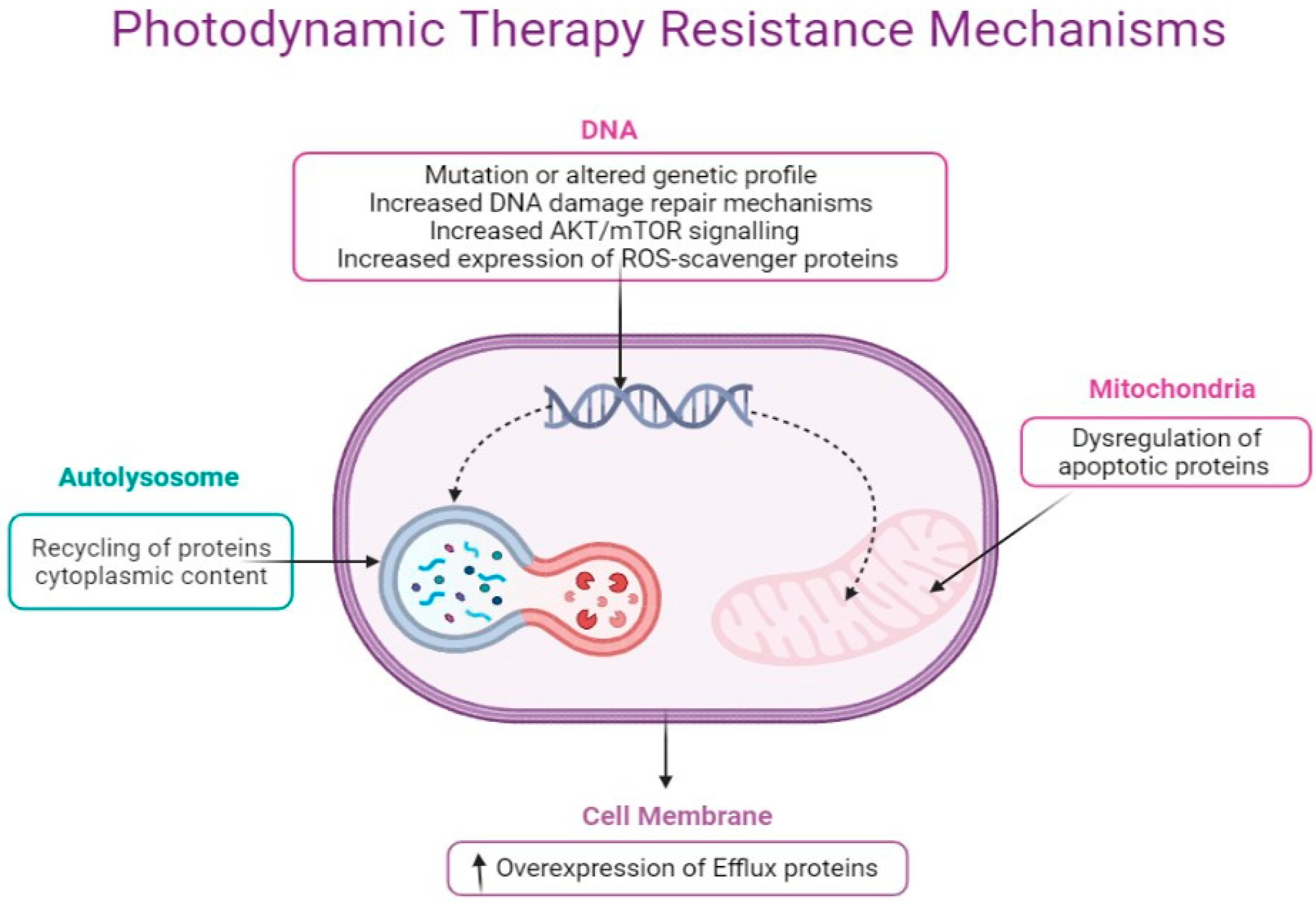
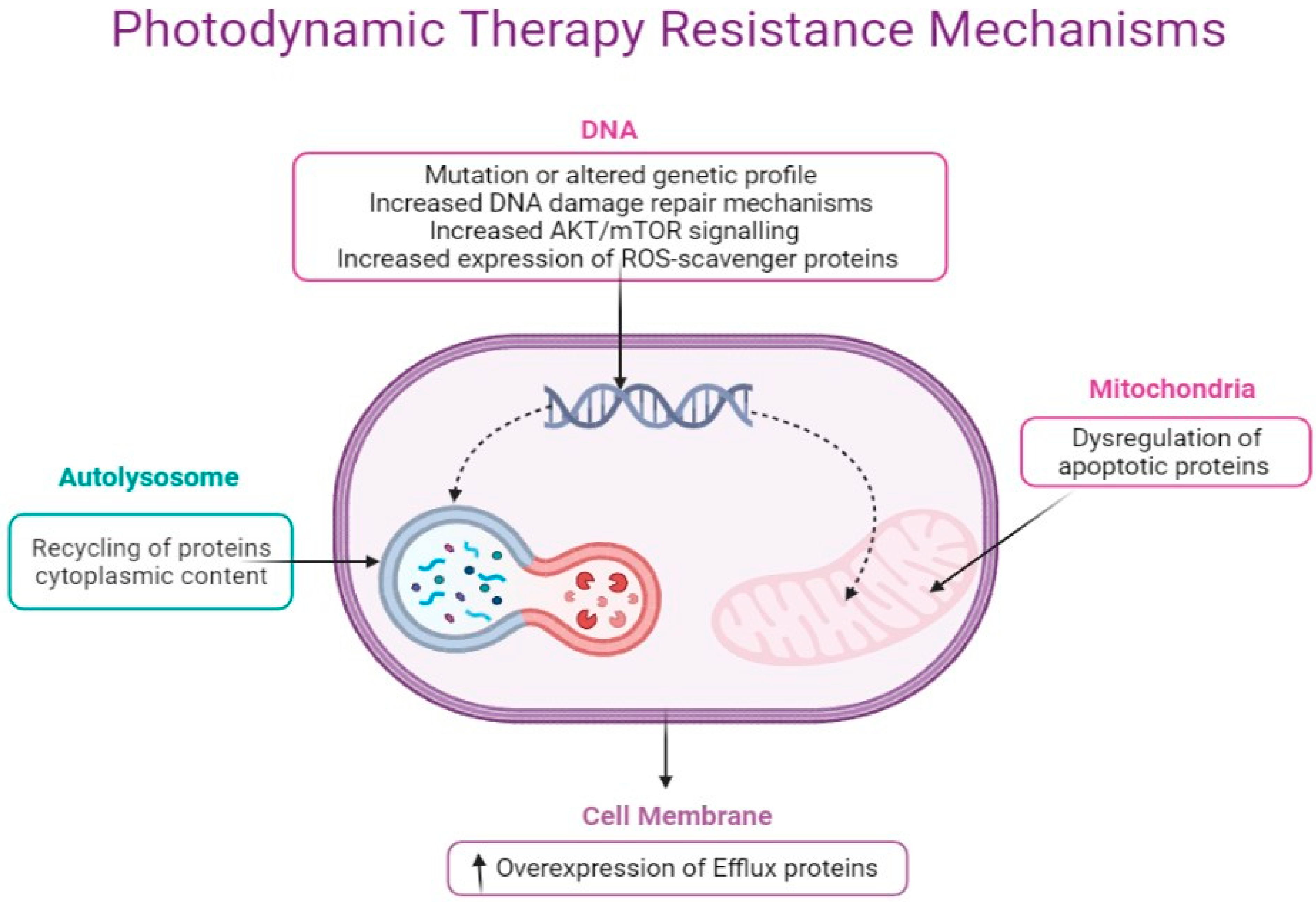
6. Molecular Mechanisms of Cancer Resistance to Photodynamic Therapy
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hönigsmann, H. History of phototherapy in dermatology. Photochem. Photobiol. Sci. 2013, 12, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Roelandts, R. The history of phototherapy: Something new under the sun? J. Am. Acad. Dermatol. 2002, 46, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Fullerenes as photosensitizers in photodynamic therapy: Pros and cons. Photochem. Photobiol. Sci. 2018, 17, 1515–1533. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Aniogo, E.C.; George, B.P.A.; Abrahamse, H. The role of photodynamic therapy on multidrug resistant breast cancer. Cancer Cell Int. 2019, 19, 91. [Google Scholar] [CrossRef]
- Vilchez, M.L.; Rodríguez, L.B.; Palacios, R.E.; Prucca, C.G.; Caverzán, M.D.; Caputto, B.L.; Rivarola, V.A.; Sanabria, L.N.M. Isolation and initial characterization of human glioblastoma cells resistant to photodynamic therapy. Photodiagnosis Photodyn. Ther. 2021, 33, 102097. [Google Scholar] [CrossRef]
- Işeri, Ö.D.; Kars, M.D.; Arpaci, F.; Gündüz, U. Gene expression analysis of drug-resistant MCF-7 cells: Implications for relation to extracellular matrix proteins. Cancer Chemother. Pharmacol. 2010, 65, 447–455. [Google Scholar] [CrossRef]
- Niculescu, A.-G.; Grumezescu, A.M. Photodynamic Therapy—An Up-to-Date Review. Appl. Sci. 2021, 11, 3626. [Google Scholar] [CrossRef]
- Baran, T. Optical Dosimetry and Treatment Planning for Photodynamic Therapy; University of Rochester: Rochester, NY, USA, 2013. [Google Scholar]
- dos Santos, A.l.F.; de Almeida, D.R.Q.; Terra, L.F.; Baptista, M.c.S.; Labriola, L. Photodynamic therapy in cancer treatment-an update review. J. Cancer Metastasis Treat. 2019, 5, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Robertson, C.A.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B Biol. 2009, 96, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Hadi, L.; MacRobert, A.J.; Loizidou, M.; Yaghini, E. Photodynamic therapy in 3D cancer models and the utilisation of nanodelivery systems. Nanoscale 2018, 10, 1570–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, C.E.; Weyergang, A.; Edwards, V.T.; Berg, K.; Brech, A.; Weisheit, S.; Høgset, A.; Selbo, P.K. Development of resistance to photodynamic therapy (pdt) in human breast cancer cells is photosensitizer-dependent: Possible mechanisms and approaches for overcoming pdt-resistance. Biochem. Pharmacol. 2017, 144, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostinis, P.; Buytaert, E.; Breyssens, H.; Hendrickx, N. Regulatory pathways in photodynamic therapy induced apoptosis. Photochem. Photobiol. Sci. 2004, 3, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Aniogo, E.C.; George, B.P.A.; Abrahamse, H. Role of Bcl-2 Family Proteins in Photodynamic Therapy Mediated Cell Survival and Regulation. Molecules 2020, 25, 5308. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Oberbauer, R. Mitochondrial regulation of apoptosis. Physiology 2003, 18, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Mroz, P.; Yaroslavsky, A.; Kharkwal, G.B.; Hamblin, M.R. Cell death pathways in photodynamic therapy of cancer. Cancers 2011, 3, 2516–2539. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, M.C.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.-F.; Chen, M.-W.; Chen, K.-C.; Lou, P.-J.; Lin, S.Y.-F.; Hung, S.-C.; Hsiao, M.; Yao, C.-J.; Shieh, M.-J. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 2014, 10, 1179–1192. [Google Scholar] [CrossRef] [Green Version]
- Broekgaarden, M.; Weijer, R.; van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015, 34, 643–690. [Google Scholar] [CrossRef] [Green Version]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar]
- Usuda, J.; Azizuddin, K.; Chiu, S.M.; Oleinick, N.L. Association Between the Photodynamic Loss of Bcl-2 and the Sensitivity to Apoptosis Caused by Phthalocyanine Photodynamic Therapy. Photochem. Photobiol. 2003, 78, 1–8. [Google Scholar] [CrossRef]
- Xue, L.; Fletcher, G.C.; Tolkovsky, A.M. Autophagy is activated by apoptotic signalling in sympathetic neurons: An alternative mechanism of death execution. Mol. Cell. Neurosci. 1999, 14, 180–198. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef] [Green Version]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. Bcl-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domagala, A.; Stachura, J.; Gabrysiak, M.; Muchowicz, A.; Zagozdzon, R.; Golab, J.; Firczuk, M. Inhibition of autophagy sensitizes cancer cells to Photofrin-based photodynamic therapy. BMC Cancer 2018, 18, 210. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewaele, M.; Martinet, W.; Rubio, N.; Verfaillie, T.; de Witte, P.A.; Piette, J.; Agostinis, P. Autophagy pathways activated in response to PDT contribute to cell resistance against ROS damage. J. Cell. Mol. Med. 2011, 15, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Fang, Z.-Z.; Zheng, Y.; Zhou, K.; Hu, C.; Krausz, K.W.; Sun, D.; Idle, J.R.; Gonzalez, F.J. Metabolic profiling of praziquantel enantiomers. Biochem. Pharmacol. 2014, 90, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wu, D.; Wang, X.; Cederbaum, A.I. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free. Radic. Biol. Med. 2012, 53, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Enriquez, S.; Kai, Y.; Maldonado, E.; Currin, R.T.; Lemasters, J.J. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy 2009, 5, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Lemasters, J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 2011, 14, 1919–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, P.D.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar]
- Zhu, J.; Tian, S.; Li, K.T.; Chen, Q.; Jiang, Y.; Lin, H.D.; Yu, L.H.; Bai, D.Q. Inhibition of breast cancer cell growth by methyl pyropheophenylchlorin photodynamic therapy is mediated though endoplasmic reticulum stress-induced autophagy in vitro and vivo. Cancer Med. 2018, 7, 1908–1920. [Google Scholar] [CrossRef]
- Huang, X.; Chen, J.; Wu, W.; Yang, W.; Zhong, B.; Qing, X.; Shao, Z. Delivery of MutT homolog 1 inhibitor by functionalized graphene oxide nanoparticles for enhanced chemo-photodynamic therapy triggers cell death in osteosarcoma. Acta Biomater. 2020, 109, 229–243. [Google Scholar] [CrossRef]
- Kong, F.; Zou, H.; Liu, X.; He, J.; Zheng, Y.; Xiong, L.; Miao, X. miR-7112-3p targets PERK to regulate the endoplasmic reticulum stress pathway and apoptosis induced by photodynamic therapy in colorectal cancer CX-1 cells. Photodiagnosis Photodyn. Ther. 2020, 29, 101663. [Google Scholar] [CrossRef]
- Zhang, L.; Ji, Z.; Zhang, J.; Yang, S. Photodynamic therapy enhances skin cancer chemotherapy effects through autophagy regulation. Photodiagnosis Photodyn. Ther. 2019, 28, 159–165. [Google Scholar] [CrossRef]
- Rosin, F.C.P.; Teixeira, M.G.; Pelissari, C.; Corrêa, L. Photodynamic Therapy Mediated by 5-aminolevulinic Acid Promotes the Upregulation and Modifies the Intracellular Expression of Surveillance Proteins in Oral Squamous Cell Carcinoma. Photochem. Photobiol. 2019, 95, 635–643. [Google Scholar] [CrossRef]
- Duan, X.; Chen, B.; Cui, Y.; Zhou, L.; Wu, C.; Yang, Z.; Wen, Y.; Miao, X.; Li, Q.; Xiong, L. Ready player one? Autophagy shapes resistance to photodynamic therapy in cancers. Apoptosis 2018, 23, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.Y.; Zacal, N.; Singh, G.; Rainbow, A.J. Alterations in Mitochondrial and Apoptosis-regulating Gene Expression in Photodynamic Therapy-resistant Variants of HT29 Colon Carcinoma Cells. Photochem. Photobiol. 2005, 81, 306–313. [Google Scholar] [CrossRef] [PubMed]
- He, G.-F.; Bian, M.-L.; Zhao, Y.-W.; Xiang, Q.; Li, H.-Y.; Xiao, C. A study on the mechanism of 5-aminolevulinic acid photodynamic therapy in vitro and in vivo in cervical cancer. Oncol. Rep. 2009, 21, 861–868. [Google Scholar] [PubMed] [Green Version]
- Chen, X.; Zhao, P.; Chen, F.; Li, L.; Luo, R. Effect and mechanism of 5-aminolevulinic acid-mediated photodynamic therapy in esophageal cancer. Lasers Med Sci. 2011, 26, 69–78. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Agarwal, M.L.; Larkin, H.E.; Friedman, L.R.; Xue, L.Y.; Olelnick, N.L. The induction of partial resistance to photodynamic therapy by the protooncogene BCL-2. Photochem. Photobiol. 1996, 64, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Yu, Q.; Huang, X.; Dai, H.; Luo, T.; Shao, J.; Chen, P.; Chen, J.; Huang, W.; Dong, X. A Highly-Efficient Type I Photosensitizer with Robust Vascular-Disruption Activity for Hypoxic-and-Metastatic Tumor Specific Photodynamic Therapy. Small 2020, 16, 2001059. [Google Scholar] [CrossRef]
- Rapozzi, V.; Jori, G. Resistance to Photodynamic Therapy in Cancer; Springer: Berlin/Heidelberg, Germany, 2014; Volume 5. [Google Scholar]
- Dong, P.; Hu, J.; Yu, S.; Zhou, Y.; Shi, T.; Zhao, Y.; Wang, X.; Liu, X. A Mitochondrial Oxidative Stress Amplifier to Overcome Hypoxia Resistance for Enhanced Photodynamic Therapy. Small Methods 2021, 5, 2100581. [Google Scholar] [CrossRef]
- Casas, A.; Di Venosa, G.; Hasan, T.; Batlle, A. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011, 18, 2486–2515. [Google Scholar] [CrossRef] [Green Version]
- Sanabria, L.M.; Rodríguez, M.E.; Cogno, I.S.; Vittar, N.B.R.; Pansa, M.F.; Lamberti, M.J.; Rivarola, V.A. Direct and indirect photodynamic therapy effects on the cellular and molecular components of the tumor microenvironment. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2013, 1835, 36–45. [Google Scholar] [CrossRef]
- Liu, C.P.; Wu, T.H.; Liu, C.Y.; Chen, K.C.; Chen, Y.X.; Chen, G.S.; Lin, S.Y. Self-supplying O2 through the catalase-like activity of gold nanoclusters for photodynamic therapy against hypoxic cancer cells. Small 2017, 13, 1700278. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, Q.; Chen, C.; Yang, G.; Cao, H.; Gao, Y.; Jiao, L.; Hao, E.; Zhang, W. NIR-absorbing superoxide radical and hyperthermia photogenerator via twisted donor-acceptor-donor molecular rotation for hypoxic tumor eradication. Sci. China Mater. 2021, 1–13. [Google Scholar] [CrossRef]
- Li, M.; Shao, Y.; Kim, J.H.; Pu, Z.; Zhao, X.; Huang, H.; Xiong, T.; Kang, Y.; Li, G.; Shao, K. Unimolecular photodynamic O2-economizer to overcome hypoxia resistance in phototherapeutics. J. Am. Chem. Soc. 2020, 142, 5380–5388. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Ma, W.; Wu, X.; Fang, W.; Guo, C.; Jin, Y. Construction of a nanotheranostic system Zr-MOF@ PPa/AF@ PEG for improved photodynamic therapy effects based on the PDT-oxygen consumption and hypoxia sensitive chemotherapeutic drug. J. Photochem. Photobiol. B Biol. 2021, 222, 112274. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Hu, F.; Duan, Y.; Wu, W.; Dong, J.; Meng, X.; Zhu, X.; Liu, B. Hybrid nanospheres to overcome hypoxia and intrinsic oxidative resistance for enhanced photodynamic therapy. ACS Nano 2020, 14, 2183–2190. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, X.; Kong, F.; Zhang, H.; Luo, L.; Li, Q.; Zhu, C.; Yang, J.; Du, Y.; You, J. Overcoming photodynamic resistance and tumor targeting dual-therapy mediated by indocyanine green conjugated gold nanospheres. J. Control. Release 2017, 258, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Zamarrón, A.; Lucena, S.R.; Salazar, N.; Sanz-Rodríguez, F.; Jaén, P.; Gilaberte, Y.; González, S.; Juarranz, Á. Isolation and characterization of PDT-resistant cancer cells. Photochem. Photobiol. Sci. 2015, 14, 1378–1389. [Google Scholar] [CrossRef]
- de Faria, C.M.G.; Inada, N.M.; Vollet-Filho, J.D.; Bagnato, V.S. A threshold dose distribution approach for the study of PDT resistance development: A threshold distribution approach for the study of PDT resistance. J. Photochem. Photobiol. B Biol. 2018, 182, 85–91. [Google Scholar] [CrossRef]
- Di Venosa, G.; Perotti, C.; Batlle, A.; Casas, A. The role of cytoskeleton and adhesion proteins in the resistance to photodynamic therapy. Possible therapeutic interventions. Photochem. Photobiol. Sci. 2015, 14, 1451–1464. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Morales Vasconsuelo, A.B.; Ferrara, M.G.; Rumie Vittar, N.B. Recapitulation of Hypoxic Tumor–stroma Microenvironment to Study Photodynamic Therapy Implications. Photochem. Photobiol. 2020, 96, 897–905. [Google Scholar] [CrossRef]
- Chizenga, E.P.; Abrahamse, H. Nanotechnology in modern photodynamic therapy of cancer: A review of cellular resistance patterns affecting the therapeutic response. Pharmaceutics 2020, 12, 632. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.E.; Jansson, P.J.; Richardson, D.R. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol. Pharmacol. 2013, 84, 655–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiya, Y.; Fukuhara, H.; Matsumoto, K.; Endo, Y.; Nakajima, M.; Tanaka, T.; Okura, I.; Kurabayashi, A.; Furihata, M.; Inoue, K. Expression levels of PEPT1 and ABCG2 play key roles in 5-aminolevulinic acid (ALA)-induced tumor-specific protoporphyrin IX (PpIX) accumulation in bladder cancer. Photodiagnosis Photodyn. Ther. 2013, 10, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Hira, D.; Terada, T. BCRP/ABCG2 and high-alert medications: Biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem. Pharmacol. 2018, 147, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kralova, J.; Kolar, M.; Kahle, M.; Truksa, J.; Lettlova, S.; Balusikova, K.; Bartunek, P. Glycol porphyrin derivatives and temoporfin elicit resistance to photodynamic therapy by different mechanisms. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jendželovský, R.; Jendželovská, Z.; Kuchárová, B.; Fedoročko, P. Breast cancer resistance protein is the enemy of hypericin accumulation and toxicity of hypericin-mediated photodynamic therapy. Biomed. Pharmacother. 2019, 109, 2173–2181. [Google Scholar] [CrossRef]
- Chekwube, A.E.; George, B.; Abrahamse, H. Phototoxic effectiveness of zinc phthalocyanine tetrasulfonic acid on MCF-7 cells with overexpressed P-glycoprotein. J. Photochem. Photobiol. B Biol. 2020, 204, 111811. [Google Scholar] [CrossRef]
- Mastrangelopoulou, M.; Grigalavicius, M.; Raabe, T.H.; Skarpen, E.; Juzenas, P.; Peng, Q.; Berg, K.; Theodossiou, T.A. Predictive biomarkers for 5-ALA-PDT can lead to personalized treatments and overcome tumor-specific resistances. Cancer Rep. 2020, 5, e1278. [Google Scholar] [CrossRef]
- Oberdanner, C.B.; Plaetzer, K.; Kiesslich, T.; Krammer, B. Photodynamic Treatment with Fractionated Light Decreases Production of Reactive Oxygen Species and Cytotoxicity In Vitro via Regeneration of Glutathione. Photochem. Photobiol. 2005, 81, 609–613. [Google Scholar] [CrossRef]
- Kim, J.; Lim, H.; Kim, S.; Cho, H.; Kim, Y.; Li, X.; Choi, H.; Kim, O. Effects of HSP27 downregulation on PDT resistance through PDT-induced autophagy in head and neck cancer cells. Oncol. Rep. 2016, 35, 2237–2245. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, H.; Lim, W.; Kim, S.; Ko, Y.; Karna, S.; Kim, O.; Choi, Y.; Choi, H.; Kim, O. Down-regulation of heat-shock protein 27–induced resistance to photodynamic therapy in oral cancer cells. J. Oral Pathol. Med. 2013, 42, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.E.; Arévalo, D.E.; Sanabria, L.M.; Carrión, F.D.C.; Fanelli, M.A.; Rivarola, V.A. Heat shock protein 27 modulates autophagy and promotes cell survival after photodynamic therapy. Photochem. Photobiol. Sci. 2019, 18, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Coupienne, I.; Bontems, S.; Dewaele, M.; Rubio, N.; Habraken, Y.; Fulda, S.; Agostinis, P.; Piette, J. NF-kappaB inhibition improves the sensitivity of human glioblastoma cells to 5-aminolevulinic acid-based photodynamic therapy. Biochem. Pharmacol. 2011, 81, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.-T.; Chien, L.-T.; Lin, Y.-H.; Chien, H.-F.; Chen, C.-T. 5-ALA mediated photodynamic therapy induces autophagic cell death via AMP-activated protein kinase. Mol. Cancer 2010, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef]
- Lin, S.; Yang, L.; Shi, H.; Du, W.; Qi, Y.; Qiu, C.; Liang, X.; Shi, W.; Liu, J. Endoplasmic reticulum-targeting photosensitizer Hypericin confers chemo-sensitization towards oxaliplatin through inducing pro-death autophagy. Int. J. Biochem. Cell Biol. 2017, 87, 54–68. [Google Scholar] [CrossRef]
- Luna, M.C.; Gomer, C.J. Isolation and initial characterization of mouse tumor cells resistant to porphyrin-mediated photodynamic therapy. Cancer Res. 1991, 51, 4243–4249. [Google Scholar]
- Lucena, S.R.; Zamarrón, A.; Carrasco, E.; Marigil, M.A.; Mascaraque, M.; Fernández-Guarino, M.; Gilaberte, Y.; González, S.; Juarranz, A. Characterisation of resistance mechanisms developed by basal cell carcinoma cells in response to repeated cycles of Photodynamic Therapy. Sci. Rep. 2019, 9, 4835. [Google Scholar] [CrossRef] [Green Version]
- Ghahe, S.S.; Kosicki, K.; Wojewódzka, M.; Majchrzak, B.A.; Fogtman, A.; Iwanicka-Nowicka, R.; Ciuba, A.; Koblowska, M.; Kruszewski, M.; Tudek, B. Increased DNA repair capacity augments resistance of glioblastoma cells to photodynamic therapy. DNA Repair 2021, 103136. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Ou, Y.; Yin, H.; Chen, Y.; Zhong, S.; Gao, Y.; Zhao, Z.; He, B.; Huang, Q.; Deng, Q. Establishment and characterization of human osteosarcoma cells resistant to pyropheophorbide-α methyl ester-mediated photodynamic therapy. Int. J. Oncol. 2017, 51, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milla, L.N.; Cogno, I.S.; Rodríguez, M.E.; Sanz-Rodríguez, F.; Zamarrón, A.; Gilaberte, Y.; Carrasco, E.; Rivarola, V.A.; Juarranz, Á. Isolation and characterization of squamous carcinoma cells resistant to photodynamic therapy. J. Cell. Biochem. 2011, 112, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Firczuk, M.; Gabrysiak, M.; Golab, J. GRP78-targeting Sensitizes Cancer Cells to Cytotoxic Effects of Photodynamic Therapy. In Resistance to Photodynamic Therapy in Cancer; Springer: Berlin/Heidelberg, Germany, 2015; pp. 149–161. [Google Scholar]
- Rivarola, V.A.; Cogno, I.S. Optimization of photodynamic therapy response by survivin gene. In Resistance to Photodynamic Therapy in Cancer; Springer: Berlin/Heidelberg, Germany, 2015; pp. 163–182. [Google Scholar]
- Toussaint, M.; Barberi-Heyob, M.; Pinel, S.; Frochot, C. How nanoparticles can solve resistance and limitation in pdt efficiency. In Resistance to Photodynamic Therapy in Cancer; Springer: Berlin/Heidelberg, Germany, 2015; pp. 197–211. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Photosensitizer | Methods Used in the Isolation of Resistant Cell Population | Cancer Cell Line Used | Features and Possible Mechanism of Resistance | References |
|---|---|---|---|---|
| Photofrin II | Short exposure (initial injury associated primarily with the plasma membrane) and long exposure to PII-PDT (associated with organelles and enzymes) damage. | RIF-1 Fibrosarcoma cells | Overlapping mechanisms of membrane-bound P-gp transport system amplification decreased DNA repair or altered biotransformation pathway. | [81] |
| Methyl-5-aminoleuvlinic acid (Me-ALA) | Red light doses and Me-ALA concentration was used after ten cycles of Me-ALA-mediated PDT. The survival criteria are PDT with a 5–15% rate. | Basal cell carcinoma | Resistance is dependent on the production of endogenous photosensitizer protoporphyrin IX and its cellular localization. | [82] |
| 5-aminolevulinic acid (5-ALA) | PDT-resistant cell line was isolated following repetitive cycles of ALA-mediated PDT. | Glioblastoma (U-87 MG) cells | High repair efficiency of oxidative DNA damage, high activity of apurinic site endonuclease 1 (APE1), and increased expression of DNA damage protein kinase ataxia telangiectasia mutated (ATM). | [83] |
| Pyropheophorbide-α methyl ester (MPPa) | Repeated cycles of PDT with increasing doses of Pyropheophorbide-α methyl ester-mediated PDT. | Human osteosarcoma (MG63 and HOS) cell lines | High expression of CD133, antiapoptotic B-cell lymphoma (Bcl-xL and Bcl-2), multidrug resistance protein 1 (MRP1), and breast cancer resistance protein (ABCG2). | [84] |
| Methyl-5-aminolevulinic acid (Me-ALA). | Successive cycles of Me-ALA-mediated PDT. Treatment conditions that caused survival rate of 5–10% were used as selection criteria. | Squamous carcinoma cells | Increased expression of cell-substrate adhesion proteins (β1-integrin, vinculin) and phosphor-survivin. | [85] |
| Methyl-5-aminolevulinic acid (Me-ALA). | The irradiation dose that caused cellular death rate of 70–90% in parental cells was selected. | Human glioblastoma cells (T98 G). | High mRNA expression levels of Fibroblastic growth factor receptor (FGFR), epidermal growth factor receptor (EGFR), and β-platelet-derived growth factor receptor (βPDGFR). | [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aniogo, E.C.; George, B.P.; Abrahamse, H. Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13182. https://doi.org/10.3390/ijms222413182
Aniogo EC, George BP, Abrahamse H. Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells. International Journal of Molecular Sciences. 2021; 22(24):13182. https://doi.org/10.3390/ijms222413182
Chicago/Turabian StyleAniogo, Eric Chekwube, Blassan P. George, and Heidi Abrahamse. 2021. "Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells" International Journal of Molecular Sciences 22, no. 24: 13182. https://doi.org/10.3390/ijms222413182
APA StyleAniogo, E. C., George, B. P., & Abrahamse, H. (2021). Molecular Effectors of Photodynamic Therapy-Mediated Resistance to Cancer Cells. International Journal of Molecular Sciences, 22(24), 13182. https://doi.org/10.3390/ijms222413182